antimicrobial_peptides_chiroptera
This repository holds fasta files and code used to predict and annotate antimicrobial peptides in Chiroptera
https://github.com/xacfran/antimicrobial_peptides_chiroptera
Science Score: 67.0%
This score indicates how likely this project is to be science-related based on various indicators:
-
✓CITATION.cff file
Found CITATION.cff file -
✓codemeta.json file
Found codemeta.json file -
✓.zenodo.json file
Found .zenodo.json file -
✓DOI references
Found 4 DOI reference(s) in README -
✓Academic publication links
Links to: frontiersin.org, zenodo.org -
○Academic email domains
-
○Institutional organization owner
-
○JOSS paper metadata
-
○Scientific vocabulary similarity
Low similarity (7.6%) to scientific vocabulary
Repository
This repository holds fasta files and code used to predict and annotate antimicrobial peptides in Chiroptera
Basic Info
- Host: GitHub
- Owner: Xacfran
- License: gpl-3.0
- Default Branch: main
- Size: 40.4 MB
Statistics
- Stars: 0
- Watchers: 1
- Forks: 0
- Open Issues: 0
- Releases: 1
Metadata Files
README.md
![]()
Antimicrobial Peptides Annotation and Prediction in Chiroptera
This project attempts to get a better understanding of the gene-family evolution of antimicrobial peptides, specifically the defensin and cathelicidin families across 20 species representing six chiropteran families. The results of this pipeline have been published in this article. Here, you will find the code used to predict and annotate antimicrobial peptides, and to identify gene gains/losses. The main resulting files of this pipeline are included in the results folder.
Table of Contents
- Antimicrobial Peptides Annotation and Prediction in Chiroptera
- Table of Contents
- Software requirements
- ORTHOLOG IDENTIFICATION IN BAT PROTEOMES
- AMP PROBABILITY ESTIMATION
- GENE SEARCH IN GENOMES
- FUNCTIONAL ANNOTATION
- FINAL AMPS PREDICTION
- PREDICTED PROTEIN COUNTS
- EXTRACT GENES FROM gffs
- ANNOTATE PROTEINS WITH DEFENSINS SUBFAMILIES
- FAMILIES EXPANSIONS AND CONTRACTIONS
- PLOTTING
- Citation
Software requirements
You should have the following software installed in your system:
The installation of MAKER2 and its dependencies is explained in the MAKER2 documentation. The installation of GMAP is detailed here. The rest of the software can be installed using conda environments.
ORTHOLOG IDENTIFICATION IN BAT PROTEOMES
Search was done using orthofisher, which initiaties a HMMER search and further incorporates a best-hit BUSCO pipeline. DEFAs with e-values < 0.001 and 80% best hits were kept, whereas 75% was used for DEFBs.
Refer to the orthofisher documentation to familiarize with the contents of fasta_args.txt used in the following loop.
```bash
for each line of the fasta_args.txt file.. do
cat $DIR/fasta_args.txt | while read LINE; do
grab the second column of the file
SPECIES=$(echo ${LINE} | awk '{print $2}')
create a directory with the name of the species
mkdir ${SPECIES}_DEFA
move into that directory
(cd ${SPECIES}_DEFA
copy files to the current directory
cp proteomes/${SPECIES}.fasta . cp defa_outgroup.hmm .
echo ${LINE} > ${SPECIES}dir.txt sed -i 's/ /\t/g' ${SPECIES}dir.txt
Activate hmmer
conda activate hmmer orthofisher -m $DIR/hmms.txt -f ${SPECIES}_dir.txt -b 0.80
Activate seqkit
conda activate seqkit seqkit fx2tab orthofisheroutput/allsequences/defaoutgroup.hmm.orthofisher -C c | awk '($4 >= 2)' | seqkit tab2fx > ${SPECIES}defa_outgroup.ortho.fa); done ``` You can either write one loop for every AMP family or write a loop that iterates through multiple AMP families.
AMP PROBABILITY ESTIMATION
After quering bat proteomes and keeping sequences based on criteria mentioned in Castellanos et al. (2023), resulting datasets were subjected to an AMP probability test using the ampir package.
First a training dataset was built following the How to train your model vignette. After the training was completed, a subsampling of the dataset (70 random sequences) is obtained and the model is tested:
```r
Create train and test sets
trainIndex <- createDataPartition(y = batsfeatures$Label, p = 0.7, list = FALSE) batsfeaturesTrain <- batsfeatures[trainIndex,] batsfeaturesTest <- bats_features[-trainIndex,]
train model
mybatsvmmodel <- train(Label~., data = batsfeaturesTrain[,-1], method ="svmRadial", trControl = trctrl_prob, preProcess = c("center", "scale"))
test model in a subsample of the dataset
testbatAMPs <- predictamps(battestset, minlen = 5, model = mybatsvm_model) ```
GENE SEARCH IN GENOMES
The AMPlify Pipeline
This workflow is taken from Li et al. 2022, and has been modified and adjusted to our needs.
Databases
Positive training set
The training set included: - APD3 database containing 3,172 AMP aminoacid sequences from a variety of Phyla. The latest version (Jan. 2020) of entire database was downloaded here. - DEFA, DEFB, and CTHL fasta files that were manually curated from NCBI and ENSEMBL of the outgroup chosen for this work. - Manually curated database of DEFA,DEFB and CTHl of bats proteomes.
After I concatenated the file that contained the bats, vertebrate and APD3 sequences (3,694 proteins), I cleaned it using seqkit as follows:
bash
seqkit fx2tab apd_bats_laura.fasta -l -i -H -C BJOUXZ | awk '{ if ($3 <= 200 && $4 < 1 ) {print $1 "\t" $2} }' | seqkit tab2fx | seqkit rmdup -s -o clean_apd_bats_laura.fasta -D duplicated_apd_bats_laura.txt
Negative training set
For the negative traning set I downloaded the Reviewed SwissProt database from here. I applied a custom script to filter the sequences using the same keywords as in the AMPlify paper
bash
grep '^AC\|^KW' uniprot_sprot.dat | awk '{printf "%s%s", /^KW/?OFS:(NR>1)?"\n":"", $0} END{print ""}' | \
sed "s/;/\t/g" | grep -iv "antimicrobial\|antibiotic\|antibacterial\|antiviral\|antifungal\|antimalarial\|antiparasitic\|anti-protist\|anticancer\|defense\|defensin\|cathelicidin\|histatin\|bacteriocin\|microbicidal\|fungicide" | \
awk -F'\t' '$3 != ""'| awk '{print $2}' > list_uniprot_non_amps.list
Potential AMPs dataset
For the potential AMPs dataset I used a variation of the code above:
bash
grep '^AC\|^KW' uniprot_sprot.dat | awk '{printf "%s%s", /^KW/?OFS:(NR>1)?"\n":"", $0} END{print ""}' | \
sed "s/;/\t/g" | grep -i "antimicrobial\|antibiotic\|antibacterial\|antiviral\|antifungal\|antimalarial\|antiparasitic\|anti-protist\|anticancer\|defense\|defensin\|cathelicidin\|histatin\|bacteriocin\|microbicidal\|fungicide" | \
awk '{print $2}' > list_uniprot_potential_amps.list
Once I got the names of the sequences that are potential AMPS, I downloaded them using this script modified from one I found online.
```bash cat listuniprotnon_amps.list | \ xargs -n 1 -P 32 -I % curl -s 'https://rest.uniprot.org/uniprotkb/search?query=%&format=fasta' \
uniprotnonamps.fasta ``` I didn´t edit the headers as in the positive training set, however, I deleted sequences that contained non-standard aminoacids (B,J,O,U,X and Z), sequences larger than 200 AAs, and lastly deleted duplicates:
bash
seqkit fx2tab uniprot_potential_amps.fasta -l -i -H -C BJOUXZ | \
awk '{ if ($3 <= 200 && $4 < 1 ) {print $1 "\t" $2} }' | seqkit tab2fx | \
seqkit rmdup -s -o clean_uniprot_potential_amps.fasta -D duplicated_potential.txt
The result is a database of 132,180 aminoacid sequences.
Aligning AMPs precursors to the genomes with GMAP
GMAP was ran in this pipeline by submitting an individual script for each species. You can replace the <NAME> of each species using a sed command and iterating the names of the species from a text file.
```bash
BAT=
Build gmap database
perl $GMAP/gmap_build -d $BAT -k 15 $GENOMES/$BAT -s none
Align cDNA from mammals to gmap database
$GMAP/gmap -d $BAT -A --max-intronlength-ends=200000 -f gff3gene -O -n 20 --nofails -B 3 -k 15 -t 10 $CDNA/mammaliacDNA_AMPs.fasta
Get scaffolds which contain hits from gmap alignment
grep -A 1000000 "gff-version" "$BAT"gmapgff3.*.out > maker/gff/"$BAT"_gmap.gff3 ```
MAKER2
Running MAKER2 is straightfoward after the installation is done. The major changes performed in this work were in the MAKER Behavior Options section in the maker_opts.ctl file, as follows:
```bash
-----MAKER Behavior Options
maxdnalen=100000 #length for dividing up contigs into chunks (increases/decreases memory usage) min_contig=1000 #skip genome contigs below this length (under 10kb are often useless)
predflank=1000 #flank for extending evidence clusters sent to gene predictors predstats=0 #report AED and QI statistics for all predictions as well as models AEDthreshold=1 #Maximum Annotation Edit Distance allowed (bound by 0 and 1) minprotein=10 #require at least this many amino acids in predicted proteins altsplice=1 #Take extra steps to try and find alternative splicing, 1 = yes, 0 = no alwayscomplete=1 #extra steps to force start and stop codons, 1 = yes, 0 = no mapforward=0 #map names and attributes forward from old GFF3 genes, 1 = yes, 0 = no keeppreds=0 #Concordance threshold to add unsupported gene prediction (bound by 0 and 1)
splithit=10000 #length for the splitting of hits (expected max intron size for evidence alignments) minintron=20 #minimum intron length (used for alignment polishing) singleexon=0 #consider single exon EST evidence when generating annotations, 1 = yes, 0 = no singlelength=250 #min length required for single exon ESTs if 'singleexon is enabled' correctest_fusion=0 #limits use of ESTs in annotation to avoid fusion genes
tries=2 #number of times to try a contig if there is a failure for some reason cleantry=0 #remove all data from previous run before retrying, 1 = yes, 0 = no cleanup=0 #removes theVoid directory with individual analysis files, 1 = yes, 0 = no TMP= #specify a directory other than the system default temporary directory for temporary files ```
Filtering MAKER results
To filter sequences with non-standard amino acids and lengths longer than 200 amino acids I used seqkit.
```bash
Conda environment
conda activate seqkit
batlistamps.txt contains the acronyms for the species i am using
for i in $(cat batlistamps.txt) do seqkit fx2tab "$i"/"$i".maker.output/"$i".all.maker.proteins.fasta -i -H -C BJOUXZ | grep -v "*" | awk '{ if ($3 < 1 ) {print} }' | seqkit tab2fx | seqkit fx2tab -l -i -H -C C | awk '{ if ($3 >= 10 && $3 <= 200 ) {print} }' | seqkit tab2fx > "$i"/"$i".maker.output/curated"$i"proteins.fasta done ```
FUNCTIONAL ANNOTATION
I used interproscan in a conda environment along with opendjk version 11 for this step. The bat_list_amps.txt in this section contains the abbreviations for the species I am working with.
```bash conda activate interpro
for i in $(cat batlistamps.txt) do interproscan.sh -i $i/"$i".maker.output/curated"$i"proteins.fasta -f tsv -dp --cpu 10 -goterms -o $i/"$i".maker.output/curated"$i"proteins.tsv done ```
Extract defensins and cathelicidins from Interpro annotation
Once Intepro was run, I extracted α-β defensins, and cathelicidins from the curated_"$i"_proteins.tsv that I got in the previous step:
```bash conda activate seqkit
for i in $(cat batlistamps.txt) do grep -i "Betadefensin|Beta-defensin" "$i"/"$i".maker.output/curated"$i"proteins.tsv | cut -f1 | uniq > "$i"/"$i".maker.output/"$i"betadefensinsheaders.txt seqtk subseq "$i"/"$i".maker.output/curated"$i"proteins.fasta "$i"/"$i".maker.output/"$i"betadefensinsheaders.txt > "$i"/"$i".maker.output/"$i"beta_defensins.fasta
Get alpha defensins files
grep -i "Alphadefensin|Alpha-defensin" "$i"/"$i".maker.output/curated"$i"proteins.tsv | cut -f1 | uniq > "$i"/"$i".maker.output/"$i"alphadefensinsheaders.txt seqtk subseq "$i"/"$i".maker.output/curated"$i"proteins.fasta "$i"/"$i".maker.output/"$i"alphadefensinsheaders.txt > "$i"/"$i".maker.output/"$i"alpha_defensins.fasta
Get cathelicidin files
grep -i "cath" "$i"/"$i".maker.output/curated"$i"proteins.tsv | cut -f1 | uniq > "$i"/"$i".maker.output/"$i"cathelicidinsheaders.txt seqtk subseq "$i"/"$i".maker.output/curated"$i"proteins.fasta "$i"/"$i".maker.output/"$i"cathelicidinsheaders.txt > "$i"/"$i".maker.output/"$i"_cathelicidins.fasta done ```
Bat defensins/cathelicidins concatenation
I ran the following command to incorporate the type of AMP and species name in the headers to make it easier for some analyses. However, I removed them later when dealing with the MAKER gff files.
```bash for i in $(cat batlistamps.txt) do
This is just for the defb, do the same for the other families
sed "s/^>/>defb"$i"/g" "$i"/"$i".maker.output/"$i"betadefensins.fasta >> bats.predicted.maker.beta.defensins.fasta done ```
Extract any other AMPs from Interpro annotation
I filtered out the defensins and cathelicidins using the headers I extracted in the previous step, from the file that contains all the proteins annotated with MAKER in each species. This will allow me to use ampir to predict which of these are in fact AMPs.
bash
for bat in $(cat bat_list_amps.txt)
do
DIR=/path/to/MAKER/results/"$bat"/"$bat".maker.output/
mkdir -p "$DIR"/all_other_amps/
cat "$DIR"/*headers.txt > "$DIR"/all_other_amps/"$bat"_def_cath_headers.txt
grep -Fvwf "$DIR"/all_other_amps/"$bat"_def_cath_headers.txt "$DIR"/curated_"$bat"_proteins.tsv | awk '{print $1}' | sort | uniq > "$DIR"/all_other_amps/"$bat"_all_other_amps_headers.txt
seqtk subseq "$DIR"/"$bat".all.maker.proteins.fasta "$DIR"/all_other_amps/"$bat"_all_other_amps_headers.txt > "$DIR"/all_other_amps/"$bat"_all_other_amps.fasta
sed -i "s/^>/"$bat"_/g" "$DIR"/all_other_amps/"$bat"_all_other_amps.fasta
done
Then I concatenated all these files into a single bats_all_other_amps.fasta file for a final prediction round.
FINAL AMPS PREDICTION
Defensins and cathelicidins prediction
To make the defensins/cathelicidins prediction of the proteins obtained in MAKER, I subsetted the potential amps dataset created here, by only selecting the defensins and cathelicidins sequences first:
bash
grep ">" uniprot_potential_amps.fasta | grep -i "cathel\|defens" | sed 's/>//g' > defens_cath_headers.txt && seqtk subseq uniprot_potential_amps.fasta defens_cath_headers.txt > uniprot_potential_defens_cath.fasta
And then I kept sequences greater than 6 AAs long:
bash
seqkit fx2tab uniprot_potential_defens_cath.fasta -l | awk '{ if ($3 >= 6 ) {print $1 "\t" $2} }' | seqkit tab2fx > uniprot_potential_defens_cath_over6aas.fasta
Now, to obtain the lengths of the sequences in the datasets I used:
bash
seqkit fx2tab uniprot_potential_defens_cath_over6aas.fasta -l | awk '{print $1"\t"$3}' > uniprot_potential_defens_cath_over6aas_length.txt
seqkit fx2tab uniprot_non_amps.fasta -l | awk '{print $1"\t"$3}' > uniprot_non_amps_length.txt
Finally, I approximated the peptide length distribution of the negative dataset to the positive dataset using the custom R script shown below.
```R
Import data
positive <- read.table("uniprotpotentialdefenscathover6aaslength.txt", header = T, sep = "\t") %>% na.omit() negative <- read.table("uniprotnonampslength.txt", header = T, sep = "\t") %>% na.omit()
Loop to approximate the negative dataset to the positive dataset
iterations <- seq(1, 210, 10) final_distribution <- c()
for (i in 1 : length(iterations)){
tmp <- negative %>% filter(length >= iterations[i] & length < iterations[i+1]) tmp <- tmp[sample(nrow(tmp), length(which(positive$length>= iterations[i] & positive$length < iterations[i+1]))), ]
message("sequence length ", iterations[i]) print(length(tmp$length)) finaldistribution <- bindrows(final_distribution,tmp) }
write.table(finaldistribution, "distributionnon_amps.txt", quote = FALSE, row.names = F, col.names = F) ``` The resulting distributions are shown below:

For alpha-beta defensins and cathelicidins prediction, I repeated the same steps as explained previously. However, I varied the dataset used as the training dataset, using the same that I built for the AMPlify pipeline, but keeping only sequences that contain more than 10 aminoacids and I fixed the length distribution as well.
I made a final multisequence alignment and got rid of sequences that did not have the 4-6 cystein motif. So the dataset went from 337 predicted proteins to 333 for the final defensins+cathelicidins dataset.
This file is found in the results folder under the name predicted_bat_defensins_cathelicidins.fasta.
Any other AMP prediction
To estimate the probability of all the other AMPs besides defensins and cathelicidins, I used the built-in predict_mature function in ampir using the bats_all_other_amps.fasta file I generated above.
```r batsallotheramps <- readfaa(file="batsallotheramps.fasta") batsallotheramps <- removenonstandardaa(batsallother_amps)
Predict with built-in model
batotherampsmature <- predictamps(batsallotheramps, model = "mature") hist(batotherampspred$prob_AMP)
Verify
batotherampsmature %>% ggplot() + geomdensity(aes(x=probAMP)) + geompoint(aes(y=0,x=prob_AMP)) + xlim(0,1)
Filter
filteredallotherampsmature <- batotherampsmature %>% filter(probAMP >= 0.8) %>% select (seqname, seqaa, prob_AMP)
Plot
filteredallotherampsmature %>% ggplot() + geomdensity(aes(x=probAMP))
Save file
dftofaa(filteredallotherampsmature, "predictedbatanyotherAMP.fasta")
``
This file is found in theresultsfolder under the namepredictedbatanyotherAMP.fasta`.
PREDICTED PROTEIN COUNTS
All other AMPs
If you want to have a detailed look at the information provided by Interpro of the proteins predicted to be AMPs by ampir, you can look at the file called all_other_amps_functional_annotation.tsv, available in the results folder. This file was obtained as follows:
```bash
Get tsv files of all the bats
for bat in $(cat batlistamps.txt) do grep ">${bat}" filtered.ampir.bats.all.other.amps.fasta | sed "s/>${bat}//g" | awk '{print $1}' >> filtered.ampir.bats.all.other.amps.headers.txt awk -v species="$bat" '{print species "\t" $0}' "$bat"/"$bat".maker.output/curated"$bat"proteins.tsv >> batscurated_proteins.tsv done
Get the annotations for the predicted proteins
grep -Fwf filtered.ampir.bats.all.other.amps.headers.txt batscuratedproteins.tsv > allotherampsfunctionalannotation.tsv ```
I also got a list of AMP names from the APD3 database to manually check and confirm for the presence of cryptic AMPs in my results.
bash
grep ">0" apd_and_vertebrates_AMPs.fasta | sed "s/|/\t/g ; s/(/\t/g; s/,/\t/g" | awk '{print $2}' | sort | uniq > list_of_APD3_amps_names.txt
Using list_of_APD3_amps_names.txt, I got rid of proteins that were not AMPs and counted predicted AMP families per species with the code below and saved it in the interpro_results.tsv file.
bash
for bat in $(cat bat_list_amps.txt)
do
column -t -s $'\t' "$bat"/"$bat".maker.output/curated_"$bat"_proteins.tsv | grep -v MobiDBLite | grep -v PRINTS | awk '{print $1 "\t" $6 "\t" $7}' | sed "s/,//g" | awk '{ if ($3 >= 0 ) {print $1 "\t" $2} }' | awk 'NR==1 {print $0}; NR>1 {if(cat[$1])cat[$1]=cat[$1]", "$2; else cat[$1]=$2;}; END{j=1; for (i in cat) print i, cat[i]}' | sed "s/^/"$bat"\t/g" | sed "s/,/\t/g" | column -t -s $'\t' | awk '{print $1 "\t" $2 "\t" $3 "\t" $4}' >> interpro.results.tsv
done
This table was used to create Fig. 1, and Table S3 of my manuscript. However, a lot of manual editing in Notepad++ and a combination of sort and uniq commands had to be applied to obtain the file called all_types_of_amps_counts.tsv that was later imported in R for plotting.
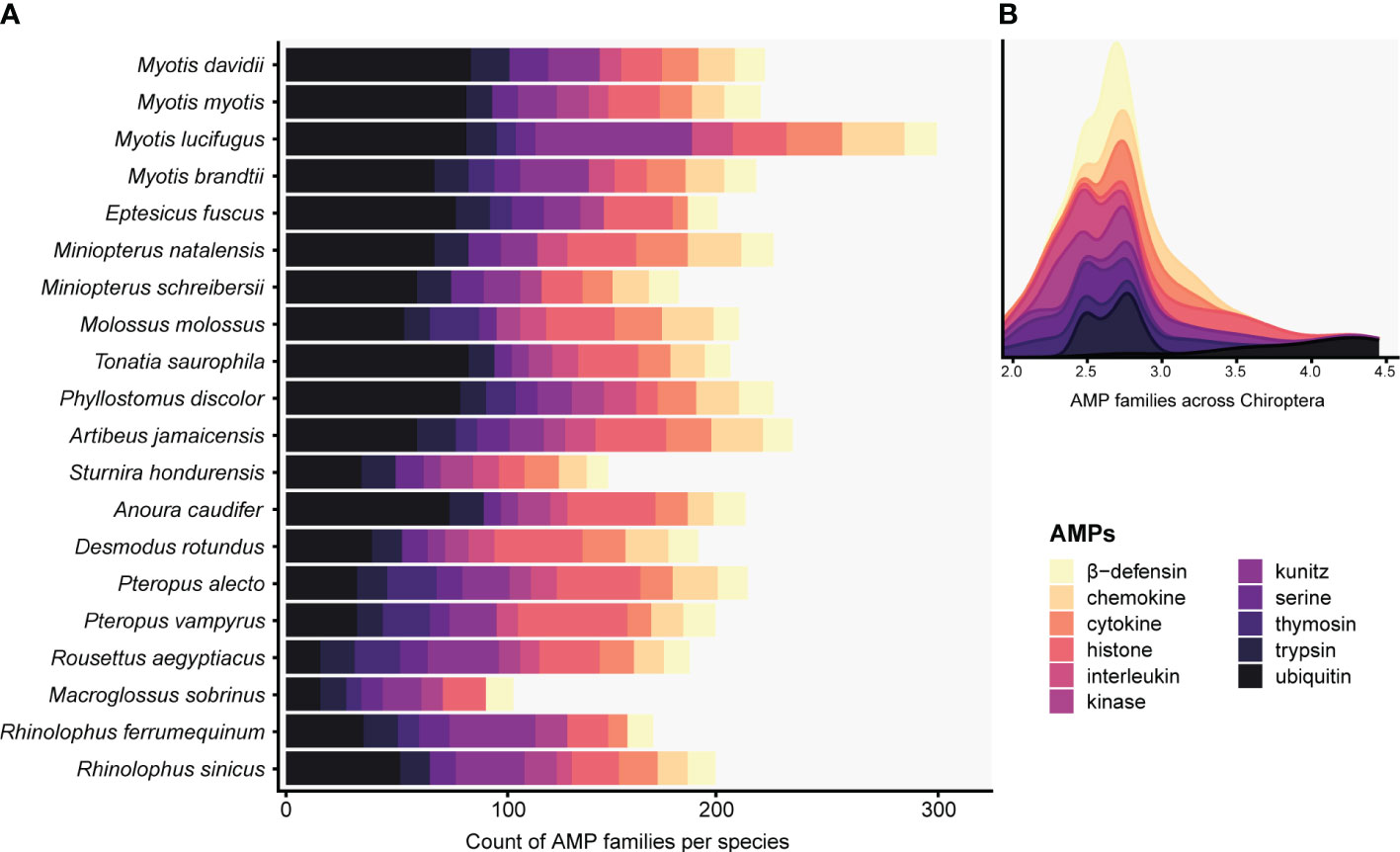{kind=link}
EXTRACT GENES FROM gffs
Extracting $\alpha$, $\beta$-defensin, and cathelicidin genes (nucleotides) from the gff files that MAKER creates was necessary for the TE analysis.
Overview stats of genes
I used the predicted_bat_defensins_cathelicidins.fasta file from the ampir prediction and the MAKER gffs to have an overview of the lengths of the genes. The bash script to estimate gene length and number of exons is shown below:
```bash
Small edits were done for the beta and cathelicidin genes
for i in $(cat batlistamps.txt)
do
while read -r line
do
genename=$(echo "$line" | awk '{print $2}')
count=$(grep "$genename" "$i".gff | awk '$3 == "exon"' | wc -l)
echo -e "$line\t$count"
done < <(grep -Fwf "$i"alphadefensinsheaders.txt "$i".gff | sed "s/=/\t/g" | awk -v species="$i" '$3 == "gene" {print species"\t"$11"\t"$5-$4}') >> batsalphadefensinsgenes_stats.txt
done
``
I had to do some manual editing in the resulting file because of a bug where some exon counts were >3 due to grep was not matching exact patterns even though I was using the-Fw` flags.
In the $\beta$-defensins I manually edited one header because of the same issue found in the $\alpha$. Also the maker-AnoCau_scaffold_2984-exonerate_protein2genome-gene-0.1 was deleted because it was not properly annotated by MAKER and had two concatenated defensin sequences in one.
Finally, for the cathelicidins I deleted one sequence because it lacked the cystein residues and another small gene of tsaurophihla.
Extract genes
The headers and the ./Gff3ToBed.sh script I created for this purpose will only retrieve the genes that are >200 bp.
```bash
!/bin/bash
Usage: ./Gff3ToBed.sh headers.txt input.gff3 output.bed
Check if input file exists
if [ ! -f $1 ] && [ -f $2 ]; then echo "Input files do not exist." exit 1 fi
Check if output file exists
if [ -f $3 ]; then echo "Output file already exists. Overwriting.." rm $3 fi
Look for the header in the gff and continue with the code
only if the gene is bigger than 200 bp.
Then print some columns of the gff and include the length of
the gene in the last column of the bed file.
grep -Fwf "$1" "$2" | awk 'BEGIN {FS="\t"; OFS="\t"} $2!~/^#/ && $3=="gene" && $5-$4>=200 { split($9,a,";"); split(a[1],b,"="); print $1,$2,$3,$4-1,$5,$6,$7,b[2],$5-$4}' > "$3.tmp"
Rename temporary file to output file
mv "$3.tmp" "$3" echo "Conversion done!" ```
Then I used bedtools to retrieve the genes from the genomes. The script requires the paths to several files created in this tutorial which will vary based on your working directory. Furthermore, the bedtools getfasta command requires the path to the softmasked genome of each species.
```bash conda activate bedtools
This is for the cathelicidins but I just had to change the names of the subfamilies accordingly
for i in $(cat batlistamps.txt) do ./Gff3ToBed.sh "$i"cathelicidinsheaders.txt "$i".gff "$i"cathelicidinsgenes.bed bedtools getfasta -fi /path/to/softmasked/genome/"$i" -bed "$i"cathelicidinsgenes.bed -fo "$i"cathelicidinsgenes.fasta done ```
ANNOTATE PROTEINS WITH DEFENSINS SUBFAMILIES
The Interpro output doesn't exactly states which defensin subfamilies I may have retrieved, thus, I blasted the dataset I used to predict AMPs in ampir to get functional names of the proteins.
First I have to copy the files to the directory and get rid of any prefix I gave to the filtered.ampir.bats.maker.predicted.amps.fasta file.
```bash
Copy files I need for this pipeline
cp /your/directory/to/filtered.ampir.bats.maker.predicted.amps.fasta . cp /your/directory/to/uniprotpotentialdefenscathover6aas.fasta .
Delete prefixes
for i in $(cat batlistamps.txt) do sed -i "s/^>defa"$i"|^>defb"$i"|^>cath"$i"/>/g" filtered.ampir.bats.maker.predicted.amps.fasta done ``` Then I blasted the potential amps file from UNIPROT that I have been using.
```bash
Create database before blasting
makeblastdb -in /your/directory/to/uniprotpotentialdefenscathover6aas.fasta -dbtype prot -out uniprotdb
BLASTP
blastp -query filtered.ampir.bats.maker.predicted.amps.fasta -db uniprotdb -evalue 1e-4 -maxhsps 1 -maxtarget_seqs 1 -outfmt 6 -out output.blastp ``` Then some looping to change the headers for the genes that were hits from BLAST.
```bash for i in $(cat batlistamps.txt) do
Get gff for the species
cp /your/directory/to/maker/"$i"/"$i".maker.output/"$i".gff .
Change headers
makermapids --prefix "$i" --justify 8 "$i".gff > "$i".contig.map
mapfastaids "$i".contig.map filtered.ampir.bats.maker.predicted.amps.fasta
Get new header names for the species
grep "$i" filtered.ampir.bats.maker.predicted.amps.fasta | sed "s/>//g" > "$i".headers.txt
Map blast results to the header
mapdataids "$i".contig.map output.blastp
Get hits from blast fot this species
grep "$i" output.blastp | sed "s/|/\t/g" | sed "s//\t/g" | cut -f1,4,6 | sed "s/\t//g" > "$i".hits.txt
Concatenate all the results
cat "$i".hits.txt >> all.hits.txt
Remove some files
rm "$i".gff "$i".contig.map "$i".headers.txt done ```
The result of this would be a change in the headers of the filtered.ampir.bats.maker.predicted.amps.fasta file. After I annotated the headers with the gene names, I had to get the genes for the outgroup and do some cleaning for < 200 AAs and excluding non traditional aminoacids.
```bash conda activate seqkit
Here I am filtering for the outgroups and get sequences using headers
for FILE in $(ls /path/to/outgroupproteins/) do grep -i "taurus|btaurus|equus|ecaballus|scrofa|sscrofa|familiaris|cfamiliaris" /path/to/outgroupproteins/$FILE >> outgroupampsheaders.txt grep -i "taurus|btaurus|equus|ecaballus|scrofa|sscrofa|familiaris|cfamiliaris" uniprotpotentialdefenscathover6aas.fasta >> outgroupampsheaders.txt sed -i "s/^>//g" outgroupampsheaders.txt seqtk subseq /path/to/outgroupproteins/$FILE outgroupampsheaders.txt >> outgroupamps.fasta done
seqtk subseq uniprotpotentialdefenscathover6aas.fasta outgroupampsheaders.txt >> outgroup_amps.fasta
Do some cleaning of the file with the mentioned criteria
seqkit fx2tab outgroupamps.fasta -l -i -H -C BJOUXZ | awk '{ if ($3 <= 200 && $4 < 1 ) {print $1 "\t" $2} }' | seqkit tab2fx | seqkit rmdup -s -o cleanoutgroupamps.fasta -D duplicatedoutgroup_amps.txt
Merge bat amps with outgroups
cat filtered.ampir.bats.maker.predicted.amps.fasta cleanoutgroupamps.fasta > outgroupbatsamps.fasta
``` After the automatic cleaning I removed some "*" that were by the end of some Equus caballus sequences only. I am reducing the complexity of the headers with some code:
bash
sed -E 's/>([a-z]+)_\S+\|(DEF[^|]+).*/>\1_\2/' outgroup_bats_amps.fasta > outgroup_bats_amps_edit_headers.fasta
sed -i "s/_Paneth_cellspecific_alphadefensin/DEFA/g ; s/_precursor//g ; s/_alphadefensin_/DEFA_/g; s/_neutrophil_defensin/DEFA/g; s/_PE=.*//g; s/OX=[*]//g" outgroup_bats_amps_edit_headers.fasta
I also had to look for the AMPs names in some sequences that were not writen in the headers, mostly sequences of cow, dog and cow corresponding to cathelicidins.
Also, since the changes of the maker_map_id get rid of the annotation of the somewhat informative headers, I am merging the results of the blast hits with the headers to make it more informative for downstream analyses.
```bash
Change the headers in outgroupbatsamps.fasta
hitsfile="all.hits.txt" fastafile="outgroupbatsampseditheaders.fasta"
Loop through each line in the hits file
while read line; do # Extract the sequence ID and desired header seqid=$(echo "$line" | cut -d"" -f1) header=$(echo "$line" | cut -d"_" -f2-)
# Use sed to replace the header in the fasta file
sed -i "s/^>$seq_id/>${seq_id}_${header}/" $fasta_file
done < $hits_file ``` Final editing to the new headers, just for simplicity
bash
sed -i "s/_Alpha-defensin//g ; s/_Defensin-.*//g ; s/_Beta-defensin//g ; s/_Cathel.*//g; s/_Defensin//g; s/_Putative//g; s/DB/DEFB/g; s/CAMP/CTHL/g; s/D103A/DEFB103A/g; s/DEF5/DEFA5/g" outgroup_bats_amps_edit_headers.fasta
I also did a manual search for the proteins that I manually separated from Phyllostomus discolor and Anoura caudifer because I added a "dup" to the header, and they were not retrieved or recognized when I ran the loop to change the header's names.
BLAST showed that both belonged to DEFB109 like proteins so I manually changed the header name to their species and DEFB, adding a "dup" at the end of it. I also had to manually look for some sequences that did not have the AMP family in them.
|Accession Number|NCBI ID | |----------------|--------| | XP024838922.1| cathelicidin-1-like | |XP024839052.1|cathelicidin-4| |XP020937599.1|protegrin-1| |NP777250.1|cathelicidin-1 precursor| |NP001123448.1|antibacterial peptide PMAP-23 precursor| |NP001123437.1|antibacterial peptide PMAP-36 precursor| |NP999615.1|antibacterial protein PR-39 precursor| |NP777250.1|cathelicidin-1 precursor| |NP777251.1|cathelicidin-2 precursor| |NP776426.1|cathelicidin-3 precursor| |NP776935.1|cathelicidin-5 precursor| |NP777256.1|cathelicidin-7 precursor| |NP777257.1|cathelicidin-6 precursor| |NP001003359.1|cathelicidin antimicrobial peptide precursor| |NP001075338.1|myeloid cathelicidin 2 precursor| |NP001075399.1|myeloid cathelicidin 3 precursor| |NP001116621.1|protegrin-1 precursor| |NP001123438.1|protegrin-2 precursor| |NP_001116622.1|protegrin-3 precursor|
FAMILIES EXPANSIONS AND CONTRACTIONS
OrthoFinder
Before running Orthofinder I had to separate the AMPs in outgroup_bats_amps_edit_headers.fasta into individual species files, including the outgroups.
```bash for i in $(cat batlistamps.txt) do
Get species headers
grep "$i" outgroupbatsampseditheaders.fasta >> ${i}ampsheaders.txt sed -i "s/^>//g" ${i}ampsheaders.txt
Get sequence
seqtk subseq outgroupbatsampseditheaders.fasta ${i}ampsheaders.txt >> ${i}.fasta rm ${i}ampsheaders.txt done
I did the same for the outgroups, however, I had to remove duplicates because, since the headers for the outgroups are similar, seqtk has difficulty in retrieving the fasta files.
```
Afterwards I ran Orthofinder within a conda environment.
bash
orthofinder -f . -t 2 -s ultrametric_tree_gh2023.tre -M msa
The results to be used for CAFE are within the Phylogenetic_Hierarchical_Orthogroups directory and I used the N0.tsv file. I did not find a way to edit the file automatically so that it can be read by mcl2rawcafe.py later. The manually edited file is in the results folder of this repository under the name hierarchical_orthogroups_edited.tsv.
CAFE5
The hierarchical_orthogroups_edited.tsv was transformed using mcl2rawcafe.py.
bash
python /path/to/cafe/mcl2rawcafe.py -i hierarchical_orthogroups_edited.tsv -o unfiltered_hierarchical_input.txt -sp "acaudifer ajamaicensis btaurus clfamiliaris drotundus ecaballus efuscus mbrandtii mdavidii mlucifugus mmolossus mmyotis mnatalensis mschreibersii msobrinus palecto pdiscolor pvampyrus raegyptiacus rferrumequinum rsinicus shondurensis sscrofa tsaurophila"
Then, two approaches were used to pick the best model. In both cases, cafe runs were done 30 times to check for the convergence of the Model Base Final Likelihood (-lnL). The highest -lnL amongst all runs was picked by checking the model_results to be submitted to a LRT.
Among gene family variation with a discrete gamma model
I picked three categories, K = 2, 3, and 5.
bash
for i in {1..30}
do
cafe5 -i unfiltered_hierarchical_input.txt -t ultrametric_tree_gh2023.tre -e -k 2 -o results_gamma_two_"$i"
done
Multi-λ model with two different birth-death parameters
For this I manually added 3 and 5 lambdas using as a guide the example of the chimphuman_separate_lambda.txt: in the CAFE5 Github repository.
Thus both of the trees written in individual files looked like:
```
lambda = 3
((((mmolossus:3,((mschreibersii:3,mnatalensis:3):3,(((mbrandtii:3,mlucifugus:3):3,(mmyotis:3,mdavidii:3):3):3,efuscus:3):3):3):3,((((shondurensis:2,ajamaicensis:2):2,(pdiscolor:2,tsaurophila:2):2):2,acaudifer:2):2,drotundus:2):2):3,((((pvampyrus:3,palecto:3):3,raegyptiacus:3):3,msobrinus:3):3,(rsinicus:3,rferrumequinum:3):3):3):1,(((btaurus:1,sscrofa:1):1,ecaballus:1):1,clfamiliaris:1):1);
lambda = 5
((((mmolossus:3,((mschreibersii:3,mnatalensis:3):3,(((mbrandtii:3,mlucifugus:3):3,(mmyotis:3,mdavidii:3):3):3,efuscus:3):3):3):3,((((shondurensis:2,ajamaicensis:2):2,(pdiscolor:2,tsaurophila:2):2):2,acaudifer:2):2,drotundus:2):2):4,((((pvampyrus:5,palecto:5):5,raegyptiacus:5):5,msobrinus:5):5,(rsinicus:5,rferrumequinum:5):5):4):1,(((btaurus:1,sscrofa:1):1,ecaballus:1):1,clfamiliaris:1):1); ```
This was used in CAFE5 as:
bash
for i in {1..30}
do
cafe5 -i unfiltered_hierarchical_input.txt -t ultrametric_tree_gh2023.tre -y ultrametric_tree_lambda_five_gh2023.txt -e -o results_lambda_five_"$i"
done
Likelihood ratio test (LRT)
To test which of the two models with highest -lnLs performed better I used R.
```r
Obtain the log-likelihoods of each model
logLik1 <- highestlnllambdacomparison logLik2 <- highestlnlkcomparison
Calculate the log-likelihood ratio
LRT <- -2*(logLik1 - logLik2)
Calculate the p-value of the LRT
pval <- pchisq(LRT, df = (5 - 3), lower.tail = FALSE) ```
PLOTTING
To be completed....
Citation
If you use this code or any dataset please cite: Castellanos FX, Moreno-Santillán D, Hughes GM, Paulat NS, Sipperly N, Brown AM, Martin KR, Poterewicz GM, Lim MCW, Russell AL, Moore MS, Johnson MG, Corthals AP, Ray DA and Da´ valos LM (2023) The evolution of antimicrobial peptides in Chiroptera. Front. Immunol. 14:1250229. doi: 10.3389/fimmu.2023.1250229
If you use this code or any dataset click in "Cite this repository" at the top of this page to get the citation in APA and BibTeX formats.
Owner
- Name: Francisco Castellanos
- Login: Xacfran
- Kind: user
- Twitter: xacfran
- Repositories: 2
- Profile: https://github.com/Xacfran
Citation (CITATION.cff)
cff-version: 1.2.0
type: dataset
message: "If you use this software, please cite it as below."
authors:
- family-names: Castellanos
given-names: Francisco X.
orcid: https://orcid.org/0000-0003-0955-8185
title: "AMPs in Chiroptera"
version: 0.2
doi: 10.5281/zenodo.8144377
date-released: 2023-09-04
url: "https://github.com/Xacfran/antimicrobial_peptides_chiroptera"
preferred-citation:
type: article
authors:
- family-names: Castellanos
given-names: Francisco X.
orcid: https://orcid.org/0000-0003-0955-8185
- family-names: Moreno-Santillan
given-names: Diana
- family-names: Hughes
given-names: Graham M.
- family-names: Paulat
given-names: Nicole S.
- family-names: Sipperly
given-names: Nicolette
- family-names: Brown
given-names: Alexis
- family-names: Martin
given-names: Katherine
- family-names: Poterewicz
given-names: Gregory M.
- family-names: Lim
given-names: Marisa C.W.
- family-names: Russell
given-names: Amy L.
- family-names: Moore
given-names: Marianne S.
- family-names: Johnson
given-names: Matthew
- family-names: Corthals
given-names: Angelique P.
- family-names: Ray
given-names: David A.
- family-names: Davalos
given-names: Liliana M.
doi: "10.3389/fimmu.2023.1250229"
journal: "Frontiers in Immunology"
start: 1250229 # First page number
title: "The evolution of antimicrobial peptides in Chiroptera"
issue: 14
year: 2023
GitHub Events
Total
- Watch event: 1
Last Year
- Watch event: 1