https://github.com/animesh/gil
Science Score: 13.0%
This score indicates how likely this project is to be science-related based on various indicators:
-
○CITATION.cff file
-
○codemeta.json file
-
○.zenodo.json file
-
✓DOI references
Found 2 DOI reference(s) in README -
○Academic publication links
-
○Academic email domains
-
○Institutional organization owner
-
○JOSS paper metadata
-
○Scientific vocabulary similarity
Low similarity (12.9%) to scientific vocabulary
Last synced: 6 months ago
·
JSON representation
Repository
Basic Info
- Host: GitHub
- Owner: animesh
- License: mit
- Default Branch: main
- Size: 642 KB
Statistics
- Stars: 0
- Watchers: 0
- Forks: 0
- Open Issues: 0
- Releases: 0
Fork of de-Boer-Lab/GIL
Created over 3 years ago
· Last pushed over 3 years ago
https://github.com/animesh/GIL/blob/main/
# GIL: Generate Indexes for Libraries GIL is a Python package that designs indexing primers for multiplexed sequencing. A web app version of GIL is available here: https://dbl-gil.streamlitapp.com/.  # Installation GIL requires Python 3.6 or higher and is not compatible with Python 2. The optional [Levenshtein](https://pypi.org/project/Levenshtein/) module (`pip install Levenshtein`) makes index generation much faster but is not required. ## Installing from GitHub To install GIL from GitHub, use `pip install git+https://github.com/de-Boer-Lab/GIL` GIL can then be run as follows: `GIL [COMMAND] [ARGUMENTS]` For help: `GIL -h` or `GIL [COMMAND] -h` See [usage examples](#usage-examples) below for more details, and the [command line arguments](#command-line-arguments) section for explanations of all arguments. ## Running from a cloned repo If you'd prefer not to install GIL, you can instead clone the repo: `git clone https://github.com/de-Boer-Lab/GIL.git` Commands can only be run from the top-level GIL directory as follows: `python -m GIL.[COMMAND] [ARGUMENTS]` For example, to generate indexes of length 10, run `python -m GIL.generate_indexes --length 10` # Introduction Multiplex sequencing allows for many samples to be sequenced in a single run, saving time and money. Unique index sequences are added to each sample library so that the sample identity of each read can be determined. Many sets of indexes are commercially available, but these become quite costly for large numbers of samples. To lower the cost of multiplex library preparation, we designed a program that generates primers containing compatible indexes, which can be ordered as standard oligos for a fraction of the cost of commercially-available indexing primers. The program outputs primer sequences in a ready-to-order format and also generates sample sheet templates for demultiplexing. ## Sequencing library structure By default, the program generates primers compatible with Illumina TruSeq. This explanation focuses on TruSeq, but note that you can generate primers for Nextera libraries or even custom library types if desired. An Illumina TruSeq dual-indexed library has the following structure (adapted from the [Teichmann Lab's GitHub](https://teichlab.github.io/scg_lib_structs/methods_html/Illumina.html), a great resource for understanding how Illumina sequencing works):5'- AATGATACGGCGACCACCGAGATCTACACNNNNNNNNACACTCTTTCCCTACACGACGCTCTTCCGATCT-insert-AGATCGGAAGAGCACACGTCTGAACTCCAGTCACNNNNNNNNATCTCGTATGCCGTCTTCTGCTTG -3' 3'- TTACTATGCCGCTGGTGGCTCTAGATGTGNNNNNNNNTGTGAGAAAGGGATGTGCTGCGAGAAGGCTAGA-insert-TCTAGCCTTCTCGTGTGCAGACTTGAGGTCAGTGNNNNNNNNTAGAGCATACGGCAGAAGACGAAC -5' Illumina P5 i5 Truseq Read 1 Truseq Read 2 i7 Illumina P7The P5 and P7 sequences bind to the flow cell, and the Read 1 and Read 2 sequences bind to sequencing primers. ## Our indexing strategy To add indexes to a library, inserts must be flanked with Read 1/Read 2 sequences. These sequences can be added using ligation: e.g. NEBNext Adaptor for Illumina, or the "stubby Y-yoke adaptor" from the [iTru Library Method](https://doi.org/10.7717/peerj.7755). These sequences can also be added by PCR for sequencing inserts from a specific locus, such as in MPRA experiments. The following diagram shows how our indexing primers anneal to a library of inserts flanked with TruSeq Read 1/Read 2 sequences. The complementary part of the primers was designed for a Tm of 65 C with Q5 polymerase.5'- AATGATACGGCGACCACCGAGATCTACACNNNNNNNNACACTCTTTCCCTACACGACG -3' 5'- ACACTCTTTCCCTACACGACGCTCTTCCGATCT-insert-AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC -3' 3'- TGTGAGAAAGGGATGTGCTGCGAGAAGGCTAGA-insert-TCTAGCCTTCTCGTGTGCAGACTTGAGGTCAGTG -5' 3'- GTGCAGACTTGAGGTCAGTGNNNNNNNNTAGAGCATACGGCAGAAGACGAAC -5' Illumina P5 i5 Truseq Read 1 Truseq Read 2 i7 Illumina P7# Command line arguments ## Arguments for index generation The following arguments can be used to customize the indexes and primers generated with the `generate_indexes` command: ### --length The length of the index. Default: 8. ### --dist The minimum Levenshtein distance between generated indexes. Also used as the minimum distance when filtering out sequences with the `--blocklist` argument. Default: 3. ### --sample-n Number of indexes to randomly sample at the start. By default, this is 5000 for index length >= 10, and for index length < 10, random sampling is not used (all possible indexes are generated at the start). Using this argument for index length < 10 will override generating all possible indexes. ### --min-GC The lower cutoff for GC content (percent). All indexes will have a strictly higher GC content. Default: 25 (CGCATATT passes, CGATATTA fails). ### --max-GC The upper cutoff for GC content (percent). All indexes will have a strictly lower GC content. Default: 75 (CGCCGATT passes, CGGCGCTA fails). ### --max-homopolymer The maximum length of homopolymer repeat that will be present in the generated indexes. Default: 2 (CCAGTTAG passes, CCCTAGAC fails). ### --max-dinu The maximum length of dinucleotide repeats that will be present in the generated indexes. Default: 2 (ATATGCAC passes, ATATATCG fails). ### --allow-start-G Allow index reads to start with G. Still filters out indexes that start with two Gs (or i5 indexes that end with two Cs). E.g. both GTATGCAC and GGTAATCG fail by default, but with this flag, GTATGCAC passes while GGTAATCG still fails. ### --no-filter-self-priming Don't filter indexes for high self-priming potential. By default, indexes with high self-priming potential are filtered out. ### --library-type Changes the type of library that primer sequences are designed for. Options: `TruSeq`, `Nextera`, or `custom`. By default the program makes TruSeq-compatible primers. If `custom` is chosen, sequences for the 5' and 3' ends of both forward and reverse primers must be supplied with the `--primer_sequences` option. ### --primer-sequences Used to supply custom primer sequences. `--library_type custom` must be chosen to use this option. Usage is `--primer_sequences ""`. For example, supplying the default TruSeq sequences: `--primer_sequences "AATGATACGGCGACCACCGAGATCTACAC ACACTCTTTCCCTACACGACG CAAGCAGAAGACGGCATACGAGAT GTGACTGGAGTTCAGACGTG"` ### --library-name The name that appears in all generated files. By default this is the same as `--library_type`, which is TruSeq by default. ### --blocklist The path to a file containing indexes that the generated indexes should be compatible with; i.e. no generated indexes will have a Levenshtein distance of less than 3 to any index in the blocklist. The file should be a plain text file with one index per line. The index sequences should be the expected index read (for i5 indexes, the expected read from MiniSeq, NextSeq, HiSeq4000, and HiSeq3000 machines). This is the **reverse complement of the index sequence present in the primer**. ### --company Format used for oligo order sheet. Different companies use different formats for their order sheets and for specifying a phosphorothioate bond. Choices: `IDT`, `Thermo`, or `Sigma`. Eurofins uses the same format as IDT. Default: `IDT`. ### --no-mod For IDT and Sigma order sheets, don't add a '*' between the last two bases of the primers in the order sheet, which specifies a phosphorothioate bond (protects primers against 3'-5' exonuclease activity of high fidelity polymerases). For Thermo, don't add the letter code for phosphorothioate bond (a different letter for each base). By default, this modification is added to primers in the order sheet. ### --seed Seed for randomly selecting compatible sequences. Setting this allows for reproducible generation of indexes. By default, sequences will be different each time the program is run (no seed). ### --out-dir The path of the directory that all files will be saved to. By default, this is "Output". ## Arguments for sample sheet creation The following arguments can be can be used when making sample sheets (e.g. for combinatorial dual index plates) with the `create_sample_sheets` command: ### --unique Create sample sheets for unique dual indexes. By default create_sample_sheets.py will create sample sheets for combinatorial dual indexes without this flag. ### --i7s **Required**. Path to a TSV file containing i7 indexes, arranged according to their position in the 96-well plate (8 rows of 12 indexes each). The sequence of the indexes should match the index sequence in the primer, **not** the sequence read during sequencing. ### --i5s **Required**. Path to a TSV file containing i5 indexes, arranged according to their position in the 96-well plate (8 rows of 12 indexes each). The sequence of the indexes should match the index sequence in the primer, **not** the sequence read during sequencing. ### --plate-name **Required**. Name of the plate. This will appear in the sample sheet filename. ### --i7-row Required unless `--unique` flag is used. The row (A-H) from the i7 plate used to make the CDI plate. ### --i5-row Required unless `--unique` flag is used. The row (A-H) from the i5 plate used to make the CDI plate. ### --out-dir The path to the directory that sample sheets will be saved to. Default is `Output/Sample_Sheets`, which is the same directory that sample sheets are saved to when generating indexes. # Overview of the index generation process ## Generate all possible indexes Our program starts by generating a list of all possible indexes of the specified length (by default, all possible 8mers). For indexes of length > 9, a random sample of indexes is used, as using all possible kmers would take too long. ## Remove starting G In sequencers that use 2-channel chemistry (e.g. NovaSeq 6000), G is not labelled with a fluorescent dye and the presence of G is inferred from a lack of signal. Illumina warns that if the first two bases in the index are both G, "signal intensity is not generated." To ensure that signal intensity is generated for every index, we remove any indexes that start with G. For the i5 (read 2) index we also remove any indexes with a reverse complement that begins with G, as this index may be read in either direction depending on the sequencer used. For a less stringent filtering, only sequences beginning with GG (and i5 ending with CC) are removed. This filtering typically results in five plates instead of three. ## Filter for GC content Indexes with extreme GC content could lead to differences in PCR efficiency between different primers. The program removes indexes with GC content less than 25% or greater than 75%. ## Remove homopolymer and dinucleotide repeats Homopolymer and dinucleotide repeats can cause issues such as polymerase stuttering and mispriming. Runs of G can also cause issues during PCR due to [intermolecular stacking](http://cshprotocols.cshlp.org/content/2009/3/pdb.tab1ip65). We decided to remove indexes with \>2 homopolymer or dinucleotide repeats to avoid these potential issues. ## Remove self-priming sequences If an index is complementary to the 3' end of the primer, the 3' end of the primer could fold back onto itself and be extended by the polymerase, ruining the primer. To prevent this issue, we compute the Hamming distance between the reverse complement of the 8 bases on the 3' end of the primer and all 8 nt windows that include the index sequence. If any of these distances are \>3, the index is filtered out. ## Select a set of dissimilar sequences After filtering, we choose a subset of the remaining sequences such that no two sequences are too similar. To calculate similarity we use [Levenshtein distance](https://en.wikipedia.org/wiki/Levenshtein_distance), which is the number of substitutions, insertions, or deletions required to change one sequence to the other sequence. We chose Levenshtein distance because it accounts for deletions, which are the most common error in synthetic oligos. The [Levenshtein module](https://pypi.org/project/Levenshtein/) `distance` function, which is implemented in C, is used by default to calculate distance. If this module isn't available, a much slower Python implementation of Levenshtein distance is used. Before selecting the set of dissimilar sequences, the indexes can optionally be filtered to remove sequences that are too similar to an existing set of indexes. For example, if you will be using these indexes alongside commercial indexes such as NEBNext Multiplex Oligos for Illumina, you can ensure that none of the generated indexes are too similar by using the `--blocklist` option along with a file containing the indexes. A more detailed explanation of this option can be found in the [command line arguments](#command-line-arguments) section. To select this dissimilar set, we first select a random sequence from the set of filtered indexes, then remove all sequences that are too similar to this sequence. We then repeat this cycle of selecting a new sequence and removing all similar sequences until there are no more indexes left to select from. This does not necessarily generate the largest possible set of sequences, but it is much faster than trying to find this optimal set of dissimilar sequences and with default settings we routinely get three 96-well plates of indexes. ## Group indexes for colour balance When only a few indexes are used in a sequencing run, colour balance must be considered. Most Illumina sequencers use one of two main technologies for imaging: 2-channel and 4-channel. A detailed explanation of why colour balance is important can be found [here](https://support-docs.illumina.com/SHARE/IndexAdapterPooling/Content/SHARE/IndexAdapterPooling/SequencingChemistry_dtP.htm). In short, during each sequencing cycle, multiple images are taken using different colours of light, as the fluorescent dyes attached to the bases are excited at different wavelengths. If all of the bases at a certain position in the indexes are imaged in the same channel, the other channel will be completely dark, which can cause the run to fail. It is therefore important that the correct diversity of bases is present at each position in the set of indexes used in a run. The program outputs indexes such that each group of four indexes along the rows of the plate is colour balanced. This allows for multiplexing of as few as four indexes without having to worry about colour balance. # Output By default, all output files are placed in a directory named `Output` at the top level of the IndexingLibrary directory. The output directory has the following structure: Output |- Order_Sheets |- Plates | |- Indexes | | |- i7 | | |- i5 | |- Primers |- Sample_sheets `Order_Sheets` contains two CSV files for each plate generated, containing the i7 and i5 primers in a format that is ready to be ordered. `Indexes` contains TSV files of indexes for each plate generated. The indexes are arranged in a standard 96-well format. Note that these are the exact sequences (5' to 3') found in the indexing primers, **not** the index sequences that will be read by the sequencer. `Primers` contains TSV files of the indexing primers for each plate generated. They are arranged identically to the index TSV files but contain the entire primer sequence, not just the index. `Sample_Sheets` contains two CSV files for each plate generated, which can be used for sample demultiplexing. Because different sequencers read the i5 index in different directions, two different sample sheets are required for each plate, named "forward" or "reverse". The forward file is used for sequencers that read the i5 index in the same direction as it appears in the primer, while the reverse file is used for sequencers that read the reverse complement of the i5 index sequence as it appears in the primer. # Unique vs Combinatorial Dual Indexes Two indexing strategies are possible with dual indexes. Unique dual indexing (UDI) uses a unique read 1 index and a unique read 2 index for each sample. As the number of samples increases, this can get expensive, as two new indexing primers are needed for each new sample. Combinatorial dual indexing (CDI) solves this problem by using indexes multiple times while ensuring that each pair of indexes is unique. Using CDI, only 20 primers are needed to index 96 samples, while 192 primers would be required with UDI. An illustration of CDI vs UDI from the Illumina website: 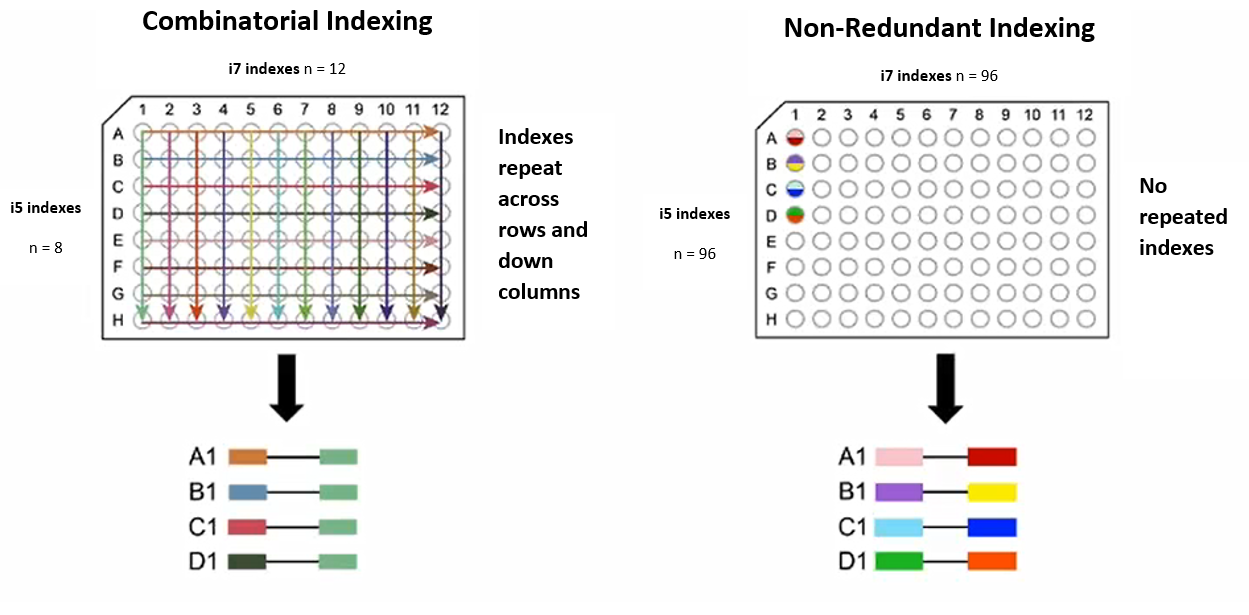 Why use UDI then? Something called [index hopping](https://www.illumina.com/techniques/sequencing/ngs-library-prep/multiplexing/index-hopping.html) can occur during sequencing, especially on certain sequencers. Index hopping is when one of the indexes from one sample occurs in a different sample. If this happens with UDIs, the index pairing will be invalid and the read can be thrown out. With CDIs, however, because indexes are shared between samples, index hopping could lead to an index combination that is still valid. This read would then be assigned to the wrong sample. ## Generating Combinatorial Index Plates By default, `generate_indexes.py` creates plates for the UDI indexing strategy: 96 i7 and 96 i5 primers for each plate generated. One set of 96 primer pairs for UDI can be expanded to 64 plates of CDI primers, which can index 6,144 samples. To do this, simply choose one row from the i7 plate and one row from the i5 plate. The 12 i7 indexes from the row are copied down the columns of the new plate, and the first 8 indexes of the i5 row are copied along the rows of the new plate. Sample sheets for a CDI plate can be created using `create_sample_sheets.py`. Specify the rows of the original UDI plates that the i7 and i5 primers were taken from, and the path to the TSV file containing the indexes for the original UDI plate. For a detailed example, see [below](#making-cdi-plates). # Usage examples ## Default settings To generate 8 bp indexes in TruSeq-compatible primers with default filtering parameters, simply run: ``` GIL generate_indexes ``` All generated files can be found in `Output` in the directory the command was run from. ## Using a blocklist Suppose you will be pooling your libraries with existing sets of indexes, for example [NEBNext Multiplex Oligos for Illumina (Dual Index Primers Set 1)](https://international.neb.com/products/e7600-nebnext-multiplex-oligos-for-illumina-dual-index-primers-set-1#Product%20Information). You can ensure that all of your generated indexes are compatible with this existing set by using the `--blocklist` option. In the manual you can find the index sequences for i7:  and i5:  Note that the expected index read is usually the reverse complement of the index sequence present in the primer, except for i5 indexes with certain sequencers. It's important that **the indexes in the blocklist are the reverse complement of the index sequence present in the primer**; i.e. the blocklist indexes should be the expected index read for MiniSeq, NextSeq, HiSeq4000, HiSeq3000. To generate indexes that are compatible with this NEB index set, create a text file (let's call it `NEB_indexes.txt`) containing one index per line: ``` AGGCTATA GCCTCTAT AGGATAGG TCAGAGCC CTTCGCCT TAAGATTA ACGTCCTG GTCAGTAC ATTACTCG TCCGGAGA CGCTCATT GAGATTCC ATTCAGAA GAATTCGT CTGAAGCT TAATGCGC CGGCTATG TCCGCGAA TCTCGCGC AGCGATAG ``` Place this file somewhere, then supply the path to this file after the `--blocklist` argument: ``` GIL generate_indexes --blocklist NEB_indexes.txt ``` All generated indexes will have a Levenshtein distance of at least 3 from any index present in the NEB index set, and libraries indexed with your generated indexes can be pooled with libraries indexed with this NEB set without index collisions. ## Using custom primer sequences You can specify custom primer sequences if you will be using the indexing primers for specialized NGS methods that don't use the standard Nextera or TruSeq adaptor sequences. Let's say you want your libraries to look like this: ``` 5'-TAAAAAAAAAACNNNNNNNNGCCCCCCCCCCA-insert-CTTTTTTTTTTANNNNNNNNACCCCCCCCCCG-3' 3'-ATTTTTTTTTTGNNNNNNNNCGGGGGGGGGGT-insert-GAAAAAAAAAATNNNNNNNNTGGGGGGGGGGC-5' idx2 (i5) read 1 read 2 idx1 (i7) ``` This means your forward/read 1/index 2/i5 primer should look like this: ``` 5'-TAAAAAAAAAACNNNNNNNNGCCCCCCCCCCA-3' ``` and your reverse/read 2/index 1/i7 primer should look like this: ``` 5'-CGGGGGGGGGGTNNNNNNNNTAAAAAAAAAAG-3' ``` To make indexing primers that will create this library structure, run the following: ``` GIL generate_indexes --library_type custom --primer_sequences "TAAAAAAAAAAC GCCCCCCCCCCA CGGGGGGGGGGT TAAAAAAAAAAG" --library_name Useless-Seq ``` The `--library_name` argument is optional and replaces "custom" in the output file names with the name of your custom library type. ## Changing index length and minimum distance between indexes By default the generated indexes have a length of 8, and the parameters for filtering were designed with this length in mind. This default length can be changed by using the `--length` argument. For indexes longer than 9 nt, a random sample of 5,000 starting indexes is used instead of using all possible indexes, as the total number of indexes possible is too large to process in a reasonable amount of time. The size of this random sample can be changed with the `--sample-n` argument. For longer indexes, you may want the minimum distance between sequences to be larger. This can be changed with the `--dist` argument. Larger distances will require a larger initial random sample to generate a sufficient number of indexes. For example, if you want indexes of length 10 with a minimum distance of 4 between any two indexes, you can run the following: ``` GIL generate_indexes --dist 4 --length 10 ``` This only generates two plates of indexes because the distance filtering is more strict. If you want more plates, you can change `--sample-n` from the default 5000: ``` GIL generate_indexes --dist 4 --length 10 --sample-n 100000 ``` This still runs in a reasonable amount of time, but now four plates of indexes are generated instead of two. ## Making CDI plates CDI plates can be created from UDI plates by copying a row of i5 primers across the rows of the CDI plate and copying a row of i7 primers down the columns of the CDI plate, as shown in the diagram:  This creates 96 unique pairs of indexes while only using 20 primers. It's important to use rows for both the i5 and i7 primers because indexes are colour balanced in groups of four across the rows of the UDI plate. If a column of i5 primers was used (which makes more sense at first glance), the CDI plate would not be guaranteed to be colour balanced. Using rows from the UDI plate ensures that the CDI plate is colour balanced in blocks of 4x4 indexes. Let's assume that you've created the CDI plate from the first i5 and i7 plates generated by GIL. To create a sample sheet for this new CDI plate, run the following: ``` GIL create_sample_sheets --i7s Output/Plates/Indexes/i7/TruSeq_i7_Indexes_Plate_1.tsv --i5s Output/Plates/Indexes/i5/TruSeq_i5_Indexes_Plate_1.tsv --i7-row A --i5-row A --plate-name CDI_plate1_i5A_i7A ``` Under `Output/Sample_Sheets` you should now see two new sample sheets: `CDI_plate1_i5A_i7A_sample_sheet_forward.csv` and `CDI_plate1_i5A_i7A_sample_sheet_reverse.csv`. Note that `--plate-name` is a required argument, but it can be whatever you want. # Testing If you've modified the code and you'd like to run our tests, run ``` python -m unittest ``` from the top level GIL directory. # Ordering primers ## Phosphorothioate modification Most commercial NGS primers contain a phosphorothioate modification between the last two nucleotides on the 3' end, which protects against primer degradation by the 3'-5' exonuclease activity of high-fidelity polymerases. By default, primers in the order sheet contain an asterisk to specify that a phosphorothioate modification should be added (this code is used by IDT, Sigma, and Eurofins). Thermo's code is different (a different letter code for each base), but you can also generate primers for ordering from Thermo by using the `--company Thermo` argument. This costs ~$4 per primer with IDT, and can be removed with the `--no-mod` option. ## Oligo quality We ordered our indexing primers as standard 100 nmol oligos from IDT, with only standard desalting for purification. Higher quality oligos, such as IDT Ultramers, will likely result in better quality sequencing data, but come at a much higher cost. We observed ~0.2% of reads with deletions in the index region for our 8 bp dual index primers, while the higher quality PhiX spike-in had only ~0.02% deletions. ## Preparing primer plates We ordered our oligos in TE buffer normalized to 100 M concentration in multiple replicate plates. We created "master plates" by diluting the i5 and i7 indexes into a single 96-well plate (polypropylene is preferable to polystyrene due to lower DNA binding) to a final concentration of 10 M each primer. We then aliquoted each master plate into multiple single-use plates to prevent index contamination between wells.
Owner
- Name: Ani
- Login: animesh
- Kind: user
- Location: Norway
- Company: Norwegian University of Science and Technology
- Website: https://www.fuzzylife.org
- Twitter: animesh1977
- Repositories: 749
- Profile: https://github.com/animesh
A medical graduate from Delhi University with post-graduation in bioinformatics from Jawaharlal Nehru University, India.