scanexitronlr
A package for the characterization and quantification of exitron splicing events in long-read RNA-seq
Science Score: 57.0%
This score indicates how likely this project is to be science-related based on various indicators:
-
✓CITATION.cff file
Found CITATION.cff file -
✓codemeta.json file
Found codemeta.json file -
✓.zenodo.json file
Found .zenodo.json file -
✓DOI references
Found 3 DOI reference(s) in README -
○Academic publication links
-
○Academic email domains
-
○Institutional organization owner
-
○JOSS paper metadata
-
○Scientific vocabulary similarity
Low similarity (16.1%) to scientific vocabulary
Keywords
Repository
A package for the characterization and quantification of exitron splicing events in long-read RNA-seq
Basic Info
- Host: GitHub
- Owner: ylab-hi
- License: mit
- Language: Python
- Default Branch: main
- Homepage: https://pypi.org/project/scanexitronlr/
- Size: 11.5 MB
Statistics
- Stars: 6
- Watchers: 3
- Forks: 0
- Open Issues: 1
- Releases: 1
Topics
Metadata Files
README.md
![]()




ScanExitronLR
A computational workflow for exitron splicing identification in long-read RNA-seq data. 

Installation
The recommended way to install ScanExitronLR is using pip:
console
$ pip install scanexitronlr
This will pull and install the latest stable release from PyPi. ScanExitronLR requires Python 3.7+. Thus you need to make sure that the pip is for python3 using e.g. which pip or using:
console
$ pip3 install scanexitronlr
To test your installation, run:
console
$ selr
You should see the version number, usage instructions and commands. (If you prefer a more descriptive command scanexitronlr also works.)
NOTE: ScanExitronLR uses the LIQA package to infer exitron specific transcript abundance. Currently, LIQA sometimes crashes with newest versions of the lifelines package. To avoid this, install version v0.26.4 of lifelines with conda install lifelines=v0.26.4.
Usage
ScanExitronLR has two modes, extract and annotate. Use extract when calling exitrons in an alignment and annotate when annotating exitrons called using extract.
Extract
`extract` requires three inputs: (1) a BAM alignment file of long-reads containing the ts:A flag (provided by default by Minimap2), (2) a reference genome and (3) a sorted and bgzip'd gene annotation file. Currently only gtf files are supported. We suggest using the `--junc-bed` parameter in minimap2 for more accurate spliced alignments. Without the parameter, it may be harder to distinguish unannoted exitron splicing events from annotated splicing events. To sort your gtf file, use the command: ```console $ awk '$1 ~ /^#/ {print $0;next} {print $0 | "sort -k1,1 -k4,4n -k5,5n"}' in.gtf > out_sorted.gtf ``` To bgzip your gene annotation file, use: ```console $ bgzip in.gtf ``` `bgzip` is part of the [htslib](http://www.htslib.org/), which you most likely already have installed if you care about BAM files. Otherwise, you can get it [here](http://www.htslib.org/). It is important to note that if you download the [latest GENCODE release](https://www.gencodegenes.org/human/) it will be in the gzip form, not bgzip. You will need to run `gzip -d` and then `bgzip`. ScanExitronLR utilizes the `gffutils` package, which requires an SQL-lite database of the annotation file. You do not need to provide such a file, as ScanExitronLR will create one if one is not found, though it may take ~20 minutes to build. It will be saved as `your_annotation.gtf.gz.db` in the same location as your annotation and will not need to be built again. In addition, we require a tabix index, and it will be created if one is not found. This should only take seconds. It will be saved as `your_annotation.gtf.gz.tbi`. Thus, if you are running ScanExitronLR on a shared server and using a shared annotation database, you may not have writing privelages in the shared space. You will need to copy your annotation file to your local directory. We have provided fully processed GTF files for [Gencode V39](https://drive.google.com/drive/folders/1LAU26BxAmTmkQdCaHJ1ba1LHBmOI8yUM?usp=sharing) and [TAIR10](https://drive.google.com/drive/folders/1FNZ5HRJOvGeiMxMObXBPgTGC2E0l3yeE?usp=sharing) for your convenience. To run ScanExitronLR in extract mode, simply run ```console $ selr extract ... ``` with the following parameters: | Parameter | Description | | --- | --- | | -i STR | REQUIRED: Input BAM file | | -g STR | REQUIRED: Input genome reference (e.g. hg38.fa) | | -r STR | REQUIRED: Input _sorted_ and bgzip'd annotation reference (e.g. gencode_v38_sorted.gtf.gz). | | -o STR | Output filename (e.g. bam_filename.exitron <- this is default) | | -a/--ao INT | Reports only exitrons with AO (# of supporting reads) of INT or above (default: 2). | | -p/--pso FLOAT | Reports only exitrons with PSO of FLOAT or above (default: 0.01). | | -c/--cores INT | Use INT cores (default: 1). Use as many as you can spare. Even large BAM files only use at most 4GB total memory on 10 cores. | | -cp/--cluster-purity FLOAT | Reports only exitrons with cluster purity of FLOAT or above (default: 0). | | -m/--mapq INT | Only considers reads with mapq score >= INT (default: 50) | | -j/--jitter INT | Treat splice-sites with fuzzy boundry of +/- INT (default: 10). | | -sr | Use this flag to skip the realignment step. | | -sa | Use this flag to save isoform abundance files for downstream differential isoform usage analysis with LIQA. Files are of the form: input.isoform.exitrons, input.isoform.normals (See example page) | We provide some functionality to perform exitron differential isoform usage with the `-sa` flag. See [here](https://github.com/ylab-hi/ScanExitronLR/tree/main/test_data) for an example.
Choosing Filtering Parameters
ScanExitronLR filters exitron splicing events based on AO (-a/--ao), PSO (-p/--pso) and cluster purity (-cp/--cluster-purity):
> __AO__. By default, ScanExitronLR only reports exitrons with at least two supporting reads (AO >= 2). This is filter out random sequencing errors that may lead to a faulty alignment and false splicing event. However, if the coverage is particularly low, you may need to set the AO threshold to 1 in order to detect exitrons in medium and lower expressed genes.
> __PSO__. By default, ScanExitronLR only reports exitrons with a splicing frequence (PSO) above 1%. Splicing events below this frequency may not be biologically relevant or may just be due to splicing noise. Setting PSO filtering to 0% is not recommended because it will increase running time and report many low quality splicing events.
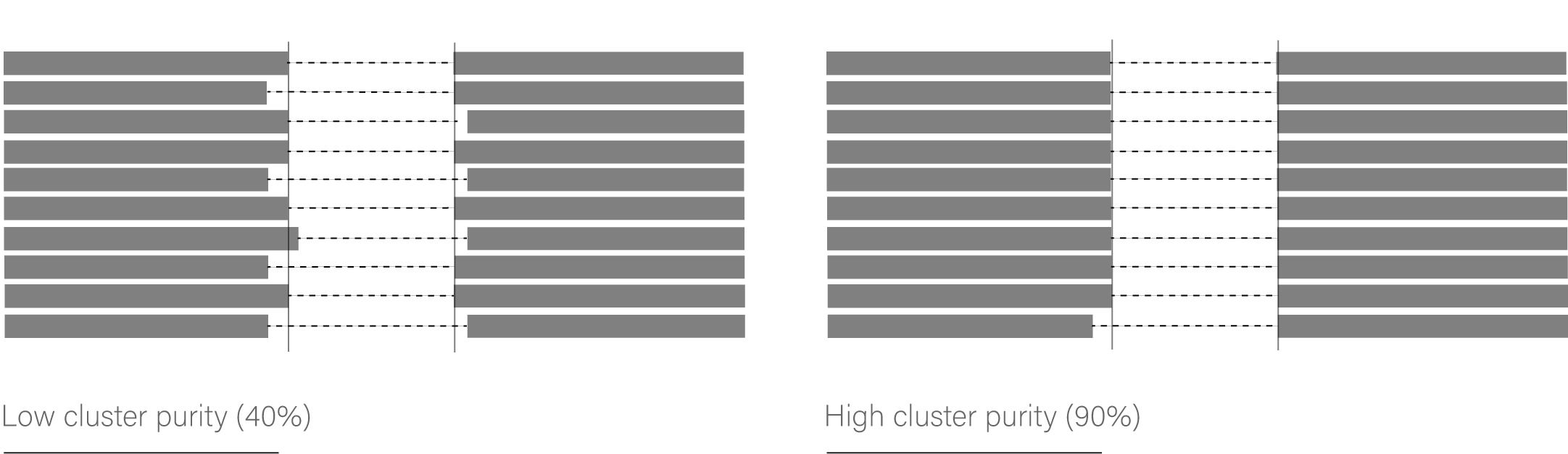
> __Cluster Purity__. By default, ScanExitronLR does not filter by cluster purity. However, cluster purity is important for having high confidence the the reported splice sites. For example, if the cluster purity is 90%, then 90% of the exitron spliced reads have the reported splice sites. Thus, one ought to be cautious when investigating exitrons with cluster purities below 50%. There is an exitron splicing event being detected, but it is unclear where the exact splice sites occur. This can happen if the reads are particularly noisy or are aligned to a repetitive region.
Annotate
To run ScanExitronLR in annotate mode, simply run
console
$ selr annotate ...
with the following parameters:
| parameters | Description | | --- | --- | | -i STR | REQUIRED: Input exitron file, generated from selr extract | | -g STR | REQUIRED: Input genome reference (e.g. hg38.fa) | | -r STR | REQUIRED: Input sorted and _ gzip'd_ annotation reference (e.g. gencodev38sorted.gtf.gz). | | -o STR | Output filename (e.g. bam_filename.exitron.annotation <- this is default) | | -b/--bam-file STR | If specified, annotation includes read supported NMD status directly from alignments. | | -arabidopsis | Use this flag if using alignments from Arabidopsis. See github page for annotation file/genome assumptions. |
The output is a tab-separated file with the following columns:
| Column | Description | | --- | --- | | chrom | Chromosome name | | start | Exitron start | | end | Exitron end | | name | Unique exitron identifier | | region | Exitron region | | ao | # of supporting reads | | strand | Gene strand | | genename | Gene name from annotation | | geneid | Gene ID from annotation | | length | Exitron legnth | | splicesite | Exitron splice sites (G[T/C]-AG, AT-AC) | | transcriptid | Transcript ID from annotation | | pso | Exitron percent spliced out value | | dp | Total depth at exitron position (PSO = AO/DP) | | clusterpurity | Exitron cluster purity | | exitronprotposition | Position in amino acid sequence of exitron splicing event | | type | Exitron type (frameshift/truncation/truncation+substitution) | | substitution | If from substitution type, determines which amino acid substitution occured | | nmdstatuspredicted | If frameshift type, determines if a downstream stop codon is 50 nt upstream of splicing junction | | nmdstatuspercentage | If frameshift type, reports percentage of reads that directly support a stop codon 50 nt upstream of splicing junction | | downstreaminframeAUG | If frameshift type, reports whether there is a downstream AUG, usually attenuating NMD efficiency | | startproximalPTC | If frameshift type, reports whether premature stop codon is within 200 nt of start codon, usually attenuating NMD efficiency | | protdomains | Any PFAM domains that are disrupted by the exitron splicing | | reads | Name of all reads which are exitron spliced |
ScanExitronLR may assign transcript abundance to multiple annotated transcripts. If this is the case, each transcript will get an annotation. Thus, if an exitron is associated with two transcripts, there will be two rows in the annotation output, one for each transcript.
Example
See here for an example.
Contact
Please feel free to post any issues here on github.
Citation
bibtex
@article{Fry2022,
doi = {10.1093/bioinformatics/btac626},
url = {https://doi.org/10.1093/bioinformatics/btac626},
year = {2022},
month = sep,
publisher = {Oxford University Press ({OUP})},
volume = {38},
number = {21},
pages = {4966--4968},
author = {Joshua Fry and Yangyang Li and Rendong Yang},
editor = {Christina Kendziorski},
title = {{ScanExitronLR}: characterization and quantification of exitron splicing events in long-read {RNA}-seq data},
journal = {Bioinformatics}
}
Owner
- Name: YangLab@Northwestern
- Login: ylab-hi
- Kind: organization
- Website: https://ylab-hi.github.io/
- Repositories: 5
- Profile: https://github.com/ylab-hi
This repo stores the codes for Yang lab projects
Citation (CITATION.cff)
cff-version: 1.2.0
message: "If you use this software, please cite it as below."
authors:
- family-names: "Fry"
given-names: "Joshua P"
orcid: "https://orcid.org/0000-0002-2503-611X"
- family-names: "Li"
given-names: "Yang Yang"
orcid: "https://orcid.org/0000-0001-8224-1067"
- family-names: "Yang"
given-names: "Ren Dong"
orcid: "https://orcid.org/0000-0001-8224-106x"
title: "ScanExitronLR: characterization and quantification of
exitron splicing events in long-read RNA-seq data"
version: 1.0.0
doi: 10.1093/bioinformatics/btac626
date-released: 2022-03-27
url: "https://github.com/ylab-hi/ScanExitronLR"
preferred-citation:
type: article
authors:
- family-names: "Fry"
given-names: "Joshua P"
orcid: "https://orcid.org/0000-0002-2503-611X"
- family-names: "Li"
given-names: "Yang Yang"
orcid: "https://orcid.org/0000-0001-8224-1067"
- family-names: "Yang"
given-names: "Ren Dong"
orcid: "https://orcid.org/0000-0001-8224-106x"
doi: "10.1093/bioinformatics/btac626"
journal: "Bioinformatics"
month: 9
start: 4966 # First page number
end: 4968 # Last page number
title: "ScanExitronLR: characterization and quantification of
exitron splicing events in long-read RNA-seq data"
issue: 21
volume: 38
year: 2022
GitHub Events
Total
Last Year
Dependencies
- pysam *