Science Score: 31.0%
This score indicates how likely this project is to be science-related based on various indicators:
-
✓CITATION.cff file
Found CITATION.cff file -
○codemeta.json file
-
○.zenodo.json file
-
✓DOI references
Found 11 DOI reference(s) in README -
○Academic publication links
-
○Academic email domains
-
○Institutional organization owner
-
○JOSS paper metadata
-
○Scientific vocabulary similarity
Low similarity (5.7%) to scientific vocabulary
Repository
Time to learn DSL2
Basic Info
Statistics
- Stars: 10
- Watchers: 2
- Forks: 4
- Open Issues: 0
- Releases: 0
Metadata Files
README.md

![]()
![]()
Introduction
nf-pcgr is a bioinformatics analysis pipeline for the functional annotation and translation of somatic SNVs/InDels and copy number abberations for precision cancer medicine using Personal Cancer Genome Reporter (PCGR). nf-pcgr offers germline SNVs/INDELS intepretation and annotation using Cancer Predisposition Sequencing Reporter (CPSR).
The workflow has been designed to accept outputs generated by nf-core/sarek:
| Tool | Germline | Somatic tumor-normal | Somatic tumor-only | | ---------------------- | :----------------: | :------------------: | :----------------: | | CNVkit | | :heavycheckmark: | :heavycheckmark: | | DeepVariant | :heavycheckmark: | | | | FreeBayes | :heavycheckmark: | :heavycheckmark: | :heavycheckmark: | | HaplotypeCaller | :heavycheckmark: | | | | Mutect2 | | :heavycheckmark: | :heavycheckmark: | | Strelka somatic indels | | :heavycheckmark: | | | Strelka somatic snvs | | :heavycheckmark: | | | Strelka variants | :heavycheckmark: | | :heavycheckmark: |
Variant consolidation
Somatic variants called by multiple tools are reformatted to match PCGR specifications making them easily searchable in the HTML ouput.
Tumor sample depth (TDP), allele frequency (TAF) and allelic depths for the ref and alt (ADT) are manually calculated and when applicable, applied to the normal sample (NDP, NAF, ADN):
```console HCC1395TvsHCC1395N.freebayes.vcf.gz
CHROM POS ID REF ALT QUAL FILTER INFO FORMAT HCC1395HCC1395N HCC1395HCC1395T
chr1 1212740 . A C 3793.78 PASS AB=0;ABP=0;AC=2;AF=0.5;AN=4;AO=126;CIGAR=1X;DP=271;DPB=271;DPRA=0.868966;EPP=5.49198;EPPR=13.9276;GTI=0;LEN=1;MEANALT=1;MQM=60;MQMR=60;NS=2;NUMALT=1;ODDS=86.3557;PAIRED=1;PAIREDR=1;PAO=0;PQA=0;PQR=0;PRO=0;QA=4420;QR=5374;RO=145;RPL=102;RPP=107.861;RPPR=111.21;RPR=24;RUN=1;SAF=49;SAP=16.5217;SAR=77;SRF=64;SRP=7.33827;SRR=81;TYPE=snp;technology.ILLUMINA=1 GT:AD:AO:DP:GQ:PL:QA:QR:RO 0/0:145,0:0:145:99:0,436,4837:0:5374:145 1/1:0,126:126:126:99:3979,379,0:4420:0:0 ```
console
TDP=126;NDP=145;TAF=1;NAF=0;ADT=0,126;ADN=145,0;TAL=freebayes
```console HCC1395TvsHCC1395N.mutect2.vcf.gz
CHROM POS ID REF ALT QUAL FILTER INFO FORMAT HCC1395HCC1395N HCC1395HCC1395T
chr1 1212740 . A C . PASS ASSBTABLE=63,80|49,76;DP=282;ECNT=1;MBQ=20,20;MFRL=151,154;MMQ=60,60;MPOS=30;NALOD=1.94;NLOD=25.89;POPAF=6.00;TLOD=341.76 GT:AD:AF:DP:F1R2:F2R1:FAD:SB 0/0:143,0:0.011:143:36,0:36,0:86,0:63,80,0,0 0/1:0,125:0.988:125:0,28:0,37:0,78:0,0,49,76 ```
console
TDP=125;NDP=143;TAF=0.988;NAF=0.011;ADT=0,125;ADN=143,0;TAL=mutect2
```console HCC1395TvsHCC1395N.strelka.somatic_snvs.vcf.gz
CHROM POS ID REF ALT QUAL FILTER INFO FORMAT NORMAL TUMOR
chr1 1212740 . A C . PASS DP=271;MQ=60.00;MQ0=0;NT=ref;QSS=790;QSSNT=3070;ReadPosRankSum=0.00;SGT=AA->AC;SNVSB=0.00;SOMATIC;SomaticEVS=19.73;TQSS=1;TQSSNT=1 DP:FDP:SDP:SUBDP:AU:CU:GU:TU 145:0:0:0:145,145:0,0:0,0:0,0 126:0:0:0:0,0:126,126:0,0:0,0 ```
console
TDP=126;NDP=145;TAF=1;NAF=0;ADT=0,126;ADN=145,0;TAL=strelka
Finally, the maximum values for TAF, TDP, NAF, NDP, ADT, ADN are taken as outputs for the consolidate variant call. In addition, values present in the ID and QUAL column (i.e not '.') are reported if present in any of the original calls:
console
1 1212740 . A C 3793.8 PASS NDP=145;NAF=0.011;TDP=126;TAF=1;TAL=freebayes,mutect2,strelka
Pipeline summary

Quick Start
Install
Nextflow(>=22.10.4)Install
DockerorSingularityDownload and unpack the human assembly-specific data bundle (grch38 for test-data):
grch37 data bundle - 20220203 (approx 20Gb)
grch38 data bundle - 20220203 (approx 21Gb)
```bash GENOME="grch38" # or "grch37" BUNDLEVERSION="20220203" BUNDLE="pcgr.databundle.${GENOME}.${BUNDLEVERSION}.tgz"
wget http://insilico.hpc.uio.no/pcgr/${BUNDLE} gzip -dc ${BUNDLE} | tar xvf - ```
Pass the directory containing the uncompressed
data/directory to nf-pcgr using the--databaseparameter for bothPCGRandCPSR.
- Download the pipeline and test it on a minimal dataset with a single command:
console
nextflow pull BarryDigby/nf-pcgr
nextflow run BarryDigby/nf-pcgr -profile test,<docker/singularity> --database '<path to PCGR database>'
Re-run the command if you encounter a
FileNotFoundError- the test data did not fully download prior to workflow execution.
Parameter documentation
Detailed descriptions of parameters can be found at parameters.md or by running nextflow run BarryDigby/nf-pcgr --help.
Usage
Input samplesheet
The workflow accepts as input a samplesheet.csv file containing the paths to SNV/InDel VCF files and CNVKit copy number abberation .cns files. We have efforted to mimick the samplesheet specifications of nf-core/sarek for ease of use:
| Column | Description | |---------|:---------------------------------------------------------------------------------------------------------------------| | patient | Designates the patient/subject; must be unique for each patient, but one patient can have multiple samples | | status | Normal/tumor (0/1) status of sample | | sample | Designates the sample ID; must be unique. A patient may have multiple samples e.g a paired tumor-normal, tumor-only. | | vcf | Full path to VCF file(s) | | cna | Full path to CNS file |
An example of a valid samplesheet is given below:
bash
patient,status,sample,vcf,cna
HCC1395,1,HCC1395T,HCC1395T_vs_HCC1395N.mutect2.vcf.gz,HCC1395T.cns
HCC1395,1,HCC1395T,HCC1395T_vs_HCC1395N.freebayes.vcf.gz,HCC1395T.cns
HCC1395,1,HCC1395T,HCC1395T_vs_HCC1395N.strelka.somatic_snvs.vcf.gz,HCC1395T.cns
HCC1395,1,HCC1395T,HCC1395T_vs_HCC1395N.strelka.somatic_indels.vcf.gz,HCC1395T.cns
HCC1395,0,HCC1395N,HCC1395N.deepvariant.vcf.gz,
HCC1395,0,HCC1395N,HCC1395N.freebayes.vcf.gz,
HCC1395,0,HCC1395N,HCC1395N.haplotypecaller.vcf.gz,
HCC1395,0,HCC1395N,HCC1395N.strelka.variants.vcf.gz,
HCC1396,1,HCC1396T,HCC1396T_vs_HCC1396N.mutect2.vcf.gz,
HCC1396,1,HCC1396T,HCC1396T_vs_HCC1396N.freebayes.vcf.gz,
HCC1396,1,HCC1396T,HCC1396T_vs_HCC1396N.strelka.somatic_snvs.vcf.gz,
HCC1396,1,HCC1396T,HCC1396T_vs_HCC1396N.strelka.somatic_indels.vcf.gz,
copy number abberation
.cnsfiles must be present for every sample entry when--cna_analysis true.
File names
Input VCF file names must contain a string denoting the variant calling tool used to detect variants between the first and second period character:
HCC1396T_vs_HCC1396N.freebayes.vcf.gz
This is the default naming convention of nf-core/sarek, thus if your VCF files originate from a different workflow you must add them prior to running nf-pcgr. Accepted strings are deepvariant, freebayes, haplotypecaller, mutect2.
For files generated by Strelka, the workflow will consider the text between the first and third period characters: strelka.variants, strelka.somatic_indels, strelka.somatic_snvs:
HCC1396T_vs_HCC1396N.strelka.somatic_indels.vcf.gz
Sample outputs
PCGR

CPSR
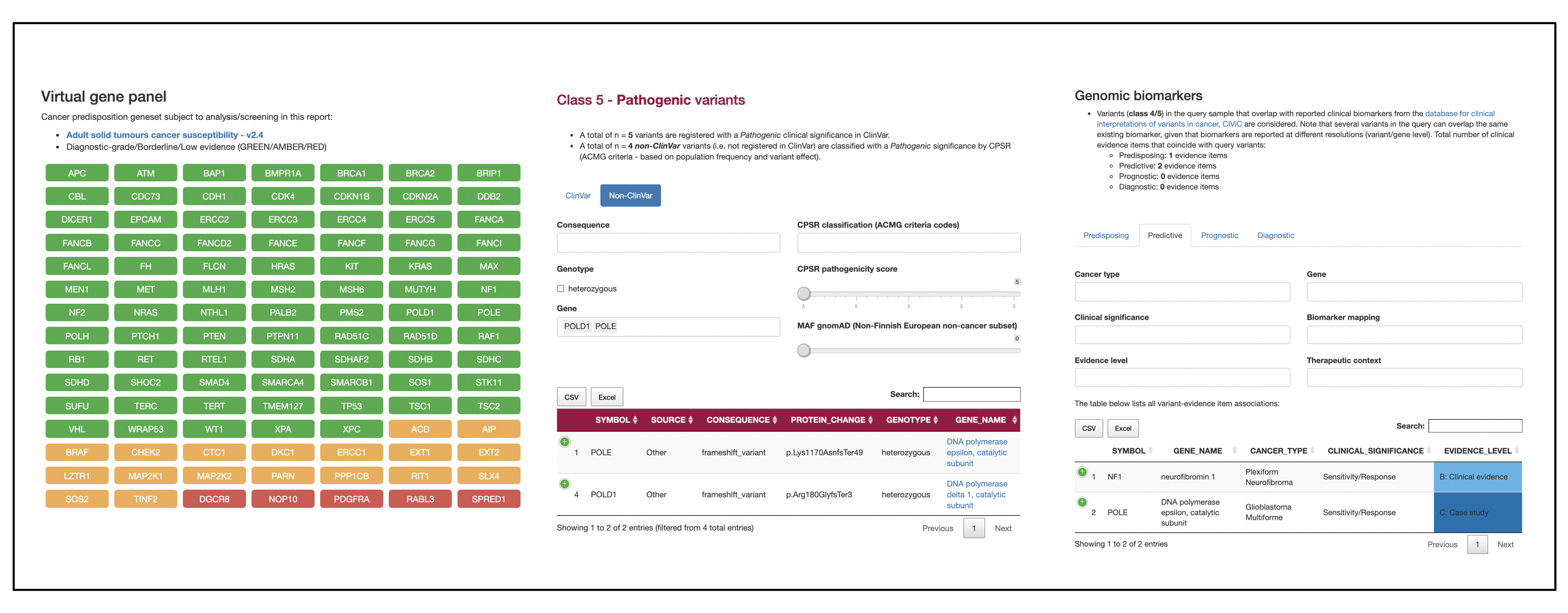
Credits
nf-core/pcgr was originally written by Barry Digby.
We thank the following people for their extensive assistance in the development of this pipeline:
Contributions and Support
Please open an issue or reach out to me (Barry Digby) on the nf-core slack channel.
I am interested in adding compatability for additional variant calling tools and optimising the intake of large VCF files.
Citations
Cancer Predisposition Sequencing Reporter (CPSR): A flexible variant report engine for high-throughput germline screening in cancer Nakken S, Saveliev V, Hofmann O, Møller P, Myklebost O, Hovig E.
Int J Cancer. 2021 Dec 1;149(11):1955-1960. doi:10.1002/ijc.33749
Personal Cancer Genome Reporter: variant interpretation report for precision oncology Nakken S, Fournous G, Vodák D, Aasheim LB, Myklebost O, Hovig E.
Bioinformatics. 2018 May 15;34(10):1778-1780. doi: 10.1093/bioinformatics/btx817
Sarek: A portable workflow for whole-genome sequencing analysis of germline and somatic variants Garcia M, Juhos S, Larsson M, Olason PI, Martin M, Eisfeldt J, DiLorenzo S, Sandgren J, Díaz De Ståhl T, Ewels P, Wirta V, Nistér M, Käller M, Nystedt B.
F1000Res. 2020 Jan 29;9:63. doi: 10.12688/f1000research.16665.2
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.
Owner
- Login: BarryDigby
- Kind: user
- Location: Galway
- Twitter: BarryDigby
- Repositories: 11
- Profile: https://github.com/BarryDigby
Repositories of a fledgling nextflow user.
Citation (CITATIONS.md)
# nf-core/pcgr: Citations ## [nf-core](https://pubmed.ncbi.nlm.nih.gov/32055031/) > Ewels PA, Peltzer A, Fillinger S, Patel H, Alneberg J, Wilm A, Garcia MU, Di Tommaso P, Nahnsen S. The nf-core framework for community-curated bioinformatics pipelines. Nat Biotechnol. 2020 Mar;38(3):276-278. doi: 10.1038/s41587-020-0439-x. PubMed PMID: 32055031. ## [Nextflow](https://pubmed.ncbi.nlm.nih.gov/28398311/) > Di Tommaso P, Chatzou M, Floden EW, Barja PP, Palumbo E, Notredame C. Nextflow enables reproducible computational workflows. Nat Biotechnol. 2017 Apr 11;35(4):316-319. doi: 10.1038/nbt.3820. PubMed PMID: 28398311. ## Pipeline tools - [FastQC](https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) - [MultiQC](https://pubmed.ncbi.nlm.nih.gov/27312411/) > Ewels P, Magnusson M, Lundin S, Käller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 2016 Oct 1;32(19):3047-8. doi: 10.1093/bioinformatics/btw354. Epub 2016 Jun 16. PubMed PMID: 27312411; PubMed Central PMCID: PMC5039924. ## Software packaging/containerisation tools - [Anaconda](https://anaconda.com) > Anaconda Software Distribution. Computer software. Vers. 2-2.4.0. Anaconda, Nov. 2016. Web. - [Bioconda](https://pubmed.ncbi.nlm.nih.gov/29967506/) > Grüning B, Dale R, Sjödin A, Chapman BA, Rowe J, Tomkins-Tinch CH, Valieris R, Köster J; Bioconda Team. Bioconda: sustainable and comprehensive software distribution for the life sciences. Nat Methods. 2018 Jul;15(7):475-476. doi: 10.1038/s41592-018-0046-7. PubMed PMID: 29967506. - [BioContainers](https://pubmed.ncbi.nlm.nih.gov/28379341/) > da Veiga Leprevost F, Grüning B, Aflitos SA, Röst HL, Uszkoreit J, Barsnes H, Vaudel M, Moreno P, Gatto L, Weber J, Bai M, Jimenez RC, Sachsenberg T, Pfeuffer J, Alvarez RV, Griss J, Nesvizhskii AI, Perez-Riverol Y. BioContainers: an open-source and community-driven framework for software standardization. Bioinformatics. 2017 Aug 15;33(16):2580-2582. doi: 10.1093/bioinformatics/btx192. PubMed PMID: 28379341; PubMed Central PMCID: PMC5870671. - [Docker](https://dl.acm.org/doi/10.5555/2600239.2600241) - [Singularity](https://pubmed.ncbi.nlm.nih.gov/28494014/) > Kurtzer GM, Sochat V, Bauer MW. Singularity: Scientific containers for mobility of compute. PLoS One. 2017 May 11;12(5):e0177459. doi: 10.1371/journal.pone.0177459. eCollection 2017. PubMed PMID: 28494014; PubMed Central PMCID: PMC5426675.
GitHub Events
Total
- Watch event: 2
- Fork event: 2
Last Year
- Watch event: 2
- Fork event: 2
Dependencies
- nf-core/tower-action v3 composite
- nf-core/tower-action v3 composite
- mshick/add-pr-comment v1 composite
- actions/checkout v3 composite
- nf-core/setup-nextflow v1 composite
- actions/checkout v3 composite
- actions/setup-node v2 composite
- actions/checkout v3 composite
- actions/setup-node v3 composite
- actions/setup-python v4 composite
- actions/upload-artifact v3 composite
- mshick/add-pr-comment v1 composite
- nf-core/setup-nextflow v1 composite
- psf/black stable composite
- dawidd6/action-download-artifact v2 composite
- marocchino/sticky-pull-request-comment v2 composite
- nfcore/base 1.14 build
- pandas *
- pysam *