crisprseq
A pipeline for the analysis of CRISPR edited data. It allows the evaluation of the quality of gene editing experiments using targeted next generation sequencing (NGS) data (`targeted`) as well as the discovery of important genes from knock-out or activation CRISPR-Cas9 screens using CRISPR pooled DNA (`screening`).
Science Score: 67.0%
This score indicates how likely this project is to be science-related based on various indicators:
-
✓CITATION.cff file
Found CITATION.cff file -
✓codemeta.json file
Found codemeta.json file -
✓.zenodo.json file
Found .zenodo.json file -
✓DOI references
Found 19 DOI reference(s) in README -
○Academic publication links
-
✓Committers with academic emails
2 of 7 committers (28.6%) from academic institutions -
○Institutional organization owner
-
○JOSS paper metadata
-
○Scientific vocabulary similarity
Low similarity (9.5%) to scientific vocabulary
Keywords
Keywords from Contributors
Repository
A pipeline for the analysis of CRISPR edited data. It allows the evaluation of the quality of gene editing experiments using targeted next generation sequencing (NGS) data (`targeted`) as well as the discovery of important genes from knock-out or activation CRISPR-Cas9 screens using CRISPR pooled DNA (`screening`).
Basic Info
- Host: GitHub
- Owner: nf-core
- License: mit
- Language: Nextflow
- Default Branch: master
- Homepage: https://nf-co.re/crisprseq
- Size: 18.8 MB
Statistics
- Stars: 47
- Watchers: 161
- Forks: 31
- Open Issues: 29
- Releases: 0
Topics
Metadata Files
README.md
![]()
![]()
![]()
![]()
![]()
Introduction
nf-core/crisprseq is a bioinformatics best-practice analysis pipeline for the analysis of CRISPR edited data. It allows the evaluation of the quality of gene editing experiments using targeted next generation sequencing (NGS) data (targeted) as well as the discovery of important genes from knock-out or activation CRISPR-Cas9 screens using CRISPR pooled DNA (screening).
nf-core/crisprseq can be used to analyse:
- CRISPR gene knockouts (KO)
- CRISPR knock-ins (KI)
- Base editing (BE) and prime editing (PE) experiments
- CRISPR screening experiments (KO, CRISPRa (activation) or CRISPRi (interference))
The pipeline is built using Nextflow, a workflow tool to run tasks across multiple compute infrastructures in a very portable manner. It uses Docker/Singularity containers making installation trivial and results highly reproducible. The Nextflow DSL2 implementation of this pipeline uses one container per process which makes it much easier to maintain and update software dependencies. Where possible, these processes have been submitted to and installed from nf-core/modules in order to make them available to all nf-core pipelines, and to everyone within the Nextflow community!
On release, automated continuous integration tests run the pipeline on a full-sized dataset on the AWS cloud infrastructure. This ensures that the pipeline runs on AWS, has sensible resource allocation defaults set to run on real-world datasets, and permits the persistent storage of results to benchmark between pipeline releases and other analysis sources. The results obtained from the full-sized test can be viewed on the nf-core website.
Pipeline summary
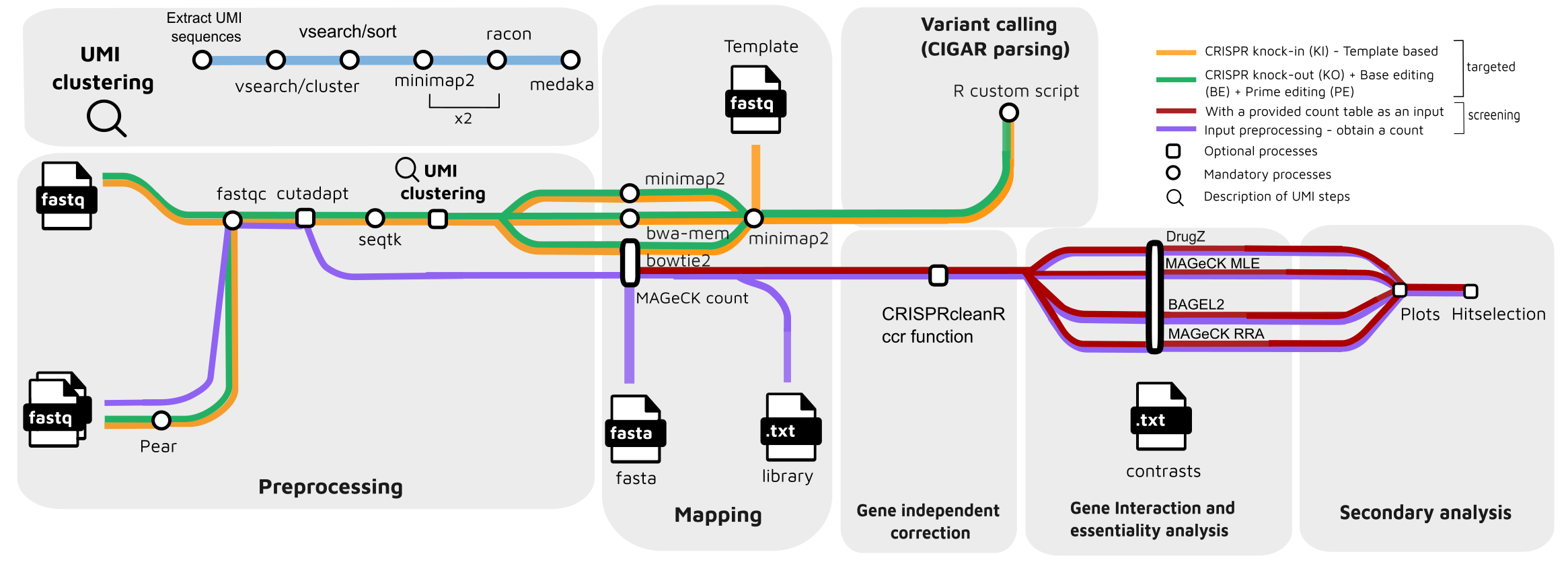
For crispr targeting :
- Merge paired-end reads (
Pear) - Read QC (
FastQC) - Adapter trimming (
Cutadapt) - Quality filtering (
Seqtk) - UMI clustering (optional):
a. Extract UMI sequences (Python script)
b. Cluster UMI sequences (
Vsearch) c. Obtain the most abundant UMI sequence for each cluster (Vsearch) d. Obtain a consensus for each cluster (minimap2) e. Polish consensus sequence (racon) f. Repeat a second rand of consensus + polishing (minimap2+racon) g. Obtain the final consensus of each cluster (Medaka) - Read mapping:
- CIGAR parsing for edit calling (
R)
For crispr screening:
- Read QC (
FastQC) - Read mapping (
MAGeCK count)- (
MAGeCK count, default) - (
bowtie2)
- (
- Optional: CNV correction and normalization with (
CRISPRcleanR) - Rank sgRNAs and genes ;
- (MAGeCK test)
- (MAGeCK mle)
- (BAGEL2)
- (DrugZ)
- Optional: hit selection on KO screen allowing a subset of more likely true positives
- Visualize analysis
Usage
[!NOTE] If you are new to Nextflow and nf-core, please refer to this page on how to set-up Nextflow. Make sure to test your setup with
-profile testbefore running the workflow on actual data.
First, prepare a samplesheet with your input data that looks as follows:
csv title="samplesheet.csv"
sample,fastq_1,fastq_2,reference,protospacer,template
SAMPLE1,SAMPLE1_R1.fastq.gz,SAMPLE1_R2.fastq.gz,ACTG,ACTG,ACTG
or
csv title="samplesheet.csv"
sample,fastq_1,fastq_2,condition
SAMPLE1,SAMPLE1_R1.fastq.gz,SAMPLE1_R2.fastq.gz,control
For more details on how to build a sample sheet, please refer to the usage documentation
Now, you can run the pipeline using:
bash
nextflow run nf-core/crisprseq --input samplesheet.csv --analysis <targeted/screening> --outdir <OUTDIR> -profile <docker/singularity/podman/shifter/charliecloud/conda/institute>
[!WARNING] Please provide pipeline parameters via the CLI or Nextflow
-params-fileoption. Custom config files including those provided by the-cNextflow option can be used to provide any configuration except for parameters; see docs.[!WARNING] Since Nextflow 23.07.0, Nextflow no longer mounts the host's HOME directory when using Apptainer or Singularity. MAGeCKFlute needs HOME directory write access. As a workaround, you can revert to the old behavior by setting the environment variable NXFAPPTAINERHOMEMOUNT or NXFSINGULARITYHOMEMOUNT to true in the machine from which you launch the pipeline.
export NXF_SINGULARITY_HOME_MOUNT=true; nextflow run nf-core/crisprseq --input samplesheet.csv --analysis screening --outdir <OUTDIR> -profile <singularity/aptainer>
For more details and further functionality, please refer to the usage documentation and the parameter documentation.
Pipeline output
To see the results of an example test run with a full size dataset refer to the results tab on the nf-core website pipeline page. For more details about the output files and reports, please refer to the output documentation.
Credits
nf-core/crisprseq targeted is based on CRISPR-A [Sanvicente-García, et.al. (2023)], originally written by Marta Sanvicente García at Translational Synthetic Biology from Universitat Pompeu Fabra. It was re-written in Nextflow DSL2 and is primarily maintained by Júlia Mir Pedrol (@mirpedrol) at Quantitative Biology Center (QBiC) from Universität Tübingen.
nf-core/crisprseq screening was written and is primarly maintained by Laurence Kuhlburger (@LaurenceKuhl) at Quantitative Biology Center (QBiC) from Universität Tübingen.
Main developers:
We thank the following people for their extensive assistance in the development of this pipeline:
- @alan-tracey
- @bolenala
- @ggabernet
- @jianhong
- @joannakraw
- @mashehu
- @medmaca
- @metinyazar
- @msanvicente
- @mschaffer-incyte
- @SusiJo
Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
For further information or help, don't hesitate to get in touch on the Slack #crisprseq channel (you can join with this invite).
Citations
If you use nf-core/crisprseq for your analysis, please cite it using the following doi: 10.5281/zenodo.7598496
An extensive list of references for the tools used by the pipeline can be found in the CITATIONS.md file.
You can cite the nf-core publication as follows:
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.
Crispr-Analytics:
Sanvicente-García M, García-Valiente A, Jouide S, Jaraba-Wallace J, Bautista E, Escobosa M, et al. (2023) CRISPR-Analytics (CRISPR-A): A platform for precise analytics and simulations for gene editing. PLoS Comput Biol 19(5): e1011137. https://doi.org/10.1371/journal.pcbi.1011137
Owner
- Name: nf-core
- Login: nf-core
- Kind: organization
- Email: core@nf-co.re
- Website: http://nf-co.re
- Twitter: nf_core
- Repositories: 84
- Profile: https://github.com/nf-core
A community effort to collect a curated set of analysis pipelines built using Nextflow.
Citation (CITATIONS.md)
# nf-core/crisprseq: Citations ## [nf-core](https://pubmed.ncbi.nlm.nih.gov/32055031/) > Ewels PA, Peltzer A, Fillinger S, Patel H, Alneberg J, Wilm A, Garcia MU, Di Tommaso P, Nahnsen S. The nf-core framework for community-curated bioinformatics pipelines. Nat Biotechnol. 2020 Mar;38(3):276-278. doi: 10.1038/s41587-020-0439-x. PubMed PMID: 32055031. ## [Nextflow](https://pubmed.ncbi.nlm.nih.gov/28398311/) > Di Tommaso P, Chatzou M, Floden EW, Barja PP, Palumbo E, Notredame C. Nextflow enables reproducible computational workflows. Nat Biotechnol. 2017 Apr 11;35(4):316-319. doi: 10.1038/nbt.3820. PubMed PMID: 28398311. ## [Crispr-Analytics](https://doi.org/10.1371/journal.pcbi.1011137) > Sanvicente-García M, García-Valiente A, Jouide S, Jaraba-Wallace J, Bautista E, Escobosa M, et al. (2023) CRISPR-Analytics (CRISPR-A): A platform for precise analytics and simulations for gene editing. PLoS Comput Biol 19(5): e1011137. https://doi.org/10.1371/journal.pcbi.1011137 ## Pipeline tools - [FastQC](https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) > Andrews, S. (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data [Online]. - [MultiQC](https://pubmed.ncbi.nlm.nih.gov/27312411/) > Ewels P, Magnusson M, Lundin S, Käller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 2016 Oct 1;32(19):3047-8. doi: 10.1093/bioinformatics/btw354. Epub 2016 Jun 16. PubMed PMID: 27312411; PubMed Central PMCID: PMC5039924. - [Pear](https://cme.h-its.org/exelixis/web/software/pear/) - [Seqtk](https://github.com/lh3/seqtk) - [Bowtie2](https://doi.org/10.1038/nmeth.1923) > Langmead B, Salzberg S. Fast gapped-read alignment with Bowtie 2. Nature Methods. 2012, 9:357-359. - [BWA](http://www.ncbi.nlm.nih.gov/pubmed/19451168) > Li H. and Durbin R. (2009) Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics, 25:1754-60. [PMID: 19451168] - [Minimap2](https://doi.org/10.1093/bioinformatics/bty191) > Li, H. (2018). Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics, 34:3094-3100. doi:10.1093/bioinformatics/bty191 - [Cutadapt](https://doi.org/10.14806/ej.17.1.200) > Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal, 17(1), pp. 10-12. doi:https://doi.org/10.14806/ej.17.1.200 - [Samtools](10.1093/bioinformatics/btp352) > Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009 Aug 15;25(16):2078-9. doi: 10.1093/bioinformatics/btp352. Epub 2009 Jun 8. PMID: 19505943; PMCID: PMC2723002. - [MAGeCKFlute](https://doi.org/10.1038/s41596-018-0113-7) > Wang, B., Wang, M., Zhang, W. et al. Integrative analysis of pooled CRISPR genetic screens using MAGeCKFlute. Nat Protoc 14, 756–780 (2019). ## Software packaging/containerisation tools - [Anaconda](https://anaconda.com) > Anaconda Software Distribution. Computer software. Vers. 2-2.4.0. Anaconda, Nov. 2016. Web. - [Bioconda](https://pubmed.ncbi.nlm.nih.gov/29967506/) > Grüning B, Dale R, Sjödin A, Chapman BA, Rowe J, Tomkins-Tinch CH, Valieris R, Köster J; Bioconda Team. Bioconda: sustainable and comprehensive software distribution for the life sciences. Nat Methods. 2018 Jul;15(7):475-476. doi: 10.1038/s41592-018-0046-7. PubMed PMID: 29967506. - [MAGeCK](https://pubmed.ncbi.nlm.nih.gov/25476604/) > Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, Liu XS. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 2014;15(12):554. - [CRISPRcleanR](https://pubmed.ncbi.nlm.nih.gov/30103702/) > Li, W. et al. Quality control, modeling, and visualization of CRISPR screens with MAGeCK-VISPR. Genome Biology 16, 281, doi:10.1186/s13059-015-0843-6 (2015). - [BAGEL2](https://pubmed.ncbi.nlm.nih.gov/33407829/) > Kim E, Hart T. Improved analysis of CRISPR fitness screens and reduced off-target effects with the BAGEL2 gene essentiality classifier. Genome Med. 2021 Jan 6;13(1):2. doi: 10.1186/s13073-020-00809-3. PMID: 33407829; PMCID: PMC7789424. - [BioContainers](https://pubmed.ncbi.nlm.nih.gov/28379341/) > da Veiga Leprevost F, Grüning B, Aflitos SA, Röst HL, Uszkoreit J, Barsnes H, Vaudel M, Moreno P, Gatto L, Weber J, Bai M, Jimenez RC, Sachsenberg T, Pfeuffer J, Alvarez RV, Griss J, Nesvizhskii AI, Perez-Riverol Y. BioContainers: an open-source and community-driven framework for software standardization. Bioinformatics. 2017 Aug 15;33(16):2580-2582. doi: 10.1093/bioinformatics/btx192. PubMed PMID: 28379341; PubMed Central PMCID: PMC5870671. - [Docker](https://dl.acm.org/doi/10.5555/2600239.2600241) > Merkel, D. (2014). Docker: lightweight linux containers for consistent development and deployment. Linux Journal, 2014(239), 2. doi: 10.5555/2600239.2600241. - [Singularity](https://pubmed.ncbi.nlm.nih.gov/28494014/) > Kurtzer GM, Sochat V, Bauer MW. Singularity: Scientific containers for mobility of compute. PLoS One. 2017 May 11;12(5):e0177459. doi: 10.1371/journal.pone.0177459. eCollection 2017. PubMed PMID: 28494014; PubMed Central PMCID: PMC5426675.
GitHub Events
Total
- Create event: 9
- Release event: 1
- Issues event: 21
- Watch event: 22
- Delete event: 20
- Issue comment event: 107
- Push event: 55
- Pull request review event: 58
- Pull request review comment event: 66
- Pull request event: 52
- Fork event: 11
Last Year
- Create event: 9
- Release event: 1
- Issues event: 21
- Watch event: 22
- Delete event: 20
- Issue comment event: 107
- Push event: 55
- Pull request review event: 58
- Pull request review comment event: 66
- Pull request event: 52
- Fork event: 11
Committers
Last synced: over 2 years ago
Top Committers
| Name | Commits | |
|---|---|---|
| mirpedrol | m****a@g****m | 208 |
| LaurenceKuhl | l****r@g****m | 55 |
| nf-core-bot | c****e@n****e | 13 |
| Julia Mir Pedrol | q****1@r****e | 3 |
| mashehu | m****3@g****m | 3 |
| marta_sanvi | m****e@u****u | 2 |
| Matthias Hörtenhuber | m****u@u****m | 1 |
Committer Domains (Top 20 + Academic)
Issues and Pull Requests
Last synced: 11 months ago
All Time
- Total issues: 34
- Total pull requests: 74
- Average time to close issues: 3 months
- Average time to close pull requests: 18 days
- Total issue authors: 13
- Total pull request authors: 8
- Average comments per issue: 1.12
- Average comments per pull request: 1.81
- Merged pull requests: 59
- Bot issues: 0
- Bot pull requests: 0
Past Year
- Issues: 14
- Pull requests: 16
- Average time to close issues: about 1 month
- Average time to close pull requests: about 1 month
- Issue authors: 10
- Pull request authors: 6
- Average comments per issue: 0.93
- Average comments per pull request: 3.38
- Merged pull requests: 8
- Bot issues: 0
- Bot pull requests: 0
Top Authors
Issue Authors
- mirpedrol (14)
- LaurenceKuhl (8)
- zhouzhendiao (4)
- matbonfanti (4)
- npatel-ah (4)
- jeremymsimon (3)
- alan-tracey (2)
- LeonHornich (2)
- dtatarak (1)
- vikramadhithyaThotam22 (1)
- KateK (1)
- tkolarik (1)
- bolenala (1)
- AASTHAPAL9 (1)
- andrewholding (1)
Pull Request Authors
- LaurenceKuhl (63)
- mirpedrol (53)
- nf-core-bot (22)
- matbonfanti (7)
- mashehu (5)
- LeonHornich (2)
- metinyazar (2)
- alan-tracey (1)
- joannakraw (1)
- mschaffer-incyte (1)
- msanvicente (1)
- jeremymsimon (1)
Top Labels
Issue Labels
Pull Request Labels
Dependencies
- actions/upload-artifact v3 composite
- nf-core/tower-action v3 composite
- actions/upload-artifact v3 composite
- nf-core/tower-action v3 composite
- mshick/add-pr-comment v1 composite
- actions/checkout v3 composite
- nf-core/setup-nextflow v1 composite
- actions/checkout v3 composite
- actions/setup-node v3 composite
- actions/checkout v3 composite
- actions/setup-node v3 composite
- actions/setup-python v4 composite
- actions/upload-artifact v3 composite
- mshick/add-pr-comment v1 composite
- nf-core/setup-nextflow v1 composite
- psf/black stable composite
- dawidd6/action-download-artifact v2 composite
- marocchino/sticky-pull-request-comment v2 composite
- actions/upload-artifact v3 composite
- nf-core/tower-action v3 composite
- actions/stale v7 composite
- actions/setup-python v4 composite
- rzr/fediverse-action master composite
- zentered/bluesky-post-action v0.0.2 composite
- bowtie2 2.4.4.*
- pigz 2.6.*
- samtools 1.16.1.*
- bowtie2 2.4.5.*
- bwa 0.7.17.*
- bwa 0.7.17.*
- samtools 1.16.1.*
- sed 4.7.*
- r-crisprcleanr 3.0.0.*
- multiqc 1.17.*
- cutadapt 3.4.*
- fastqc 0.12.1.*
- mageck 0.5.9.*
- mageck 0.5.9.*
- mageck 0.5.9.*
- medaka 1.4.4.*
- minimap2 2.24.*
- samtools 1.14.*
- minimap2 2.24.*
- multiqc 1.17.*
- pear 0.9.6.*
- racon 1.4.20.*
- samtools 1.17.*
- seqtk 1.4.*
- samtools 1.16.1.*
- vsearch 2.21.1.*
- vsearch 2.21.1.*