rsvrecon
A pipeline for assembling genomic sequences of respiratory syncytial virus (RSV) from NGS data
Science Score: 49.0%
This score indicates how likely this project is to be science-related based on various indicators:
-
○CITATION.cff file
-
✓codemeta.json file
Found codemeta.json file -
✓.zenodo.json file
Found .zenodo.json file -
✓DOI references
Found 4 DOI reference(s) in README -
✓Academic publication links
Links to: biorxiv.org, ncbi.nlm.nih.gov -
○Academic email domains
-
○Institutional organization owner
-
○JOSS paper metadata
-
○Scientific vocabulary similarity
Low similarity (8.7%) to scientific vocabulary
Repository
A pipeline for assembling genomic sequences of respiratory syncytial virus (RSV) from NGS data
Basic Info
- Host: GitHub
- Owner: stjudecab
- License: mit
- Language: Python
- Default Branch: main
- Size: 53 MB
Statistics
- Stars: 1
- Watchers: 2
- Forks: 0
- Open Issues: 1
- Releases: 0
Metadata Files
README.md
stjudecab/rsvrecon





![]()
![]()

News and Updates
- [2025-06-09] :fire: stjudecab/rsvrecon v0.1 is released.
Introduction
stjudecab/rsvrecon is a bioinformatics workflow developed to assemble and analyze genomic sequences of Respiratory Syncytial Virus (RSV) from Next-Generation Sequencing (NGS) data. It identifies genomic variations within RSV samples and highlights clinically relevant genomic features. To simplify interpretation, the workflow generates easy-to-understand HTML and PDF reports summarizing the results.
Built using Nextflow, the pipeline offers scalability, portability, and reproducibility
across diverse computational infrastructures. Dependency management is simplified by employing containerization
technologies such as Docker, Singularity, and Conda.
This pipeline utilizes the Nextflow DSL2 framework, featuring modularized processes with independent software environments, thereby making updates and maintenance straightforward. Processes are also integrated, whenever feasible, with the nf-core/modules repository to enhance usability and foster community contributions.
The schematic overview of the stjudecab/rsvrecon workflow is shown below:
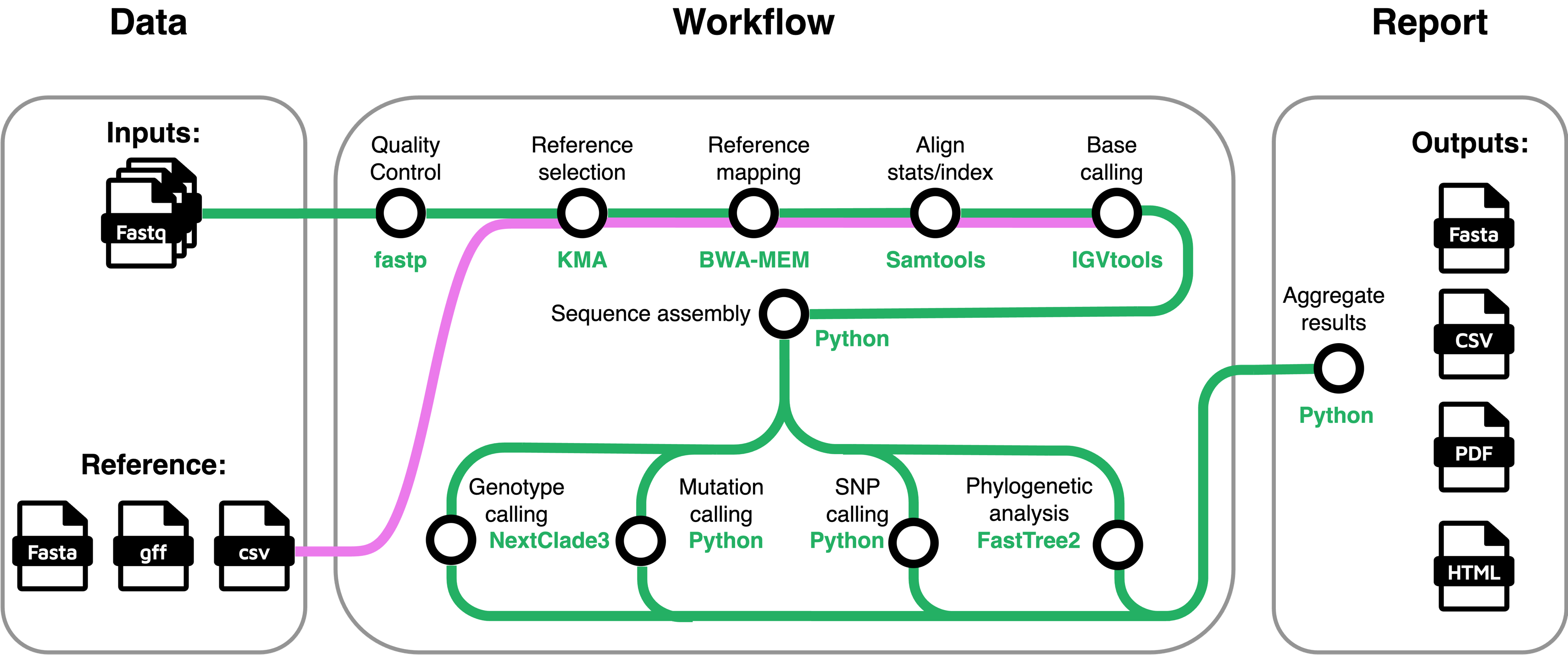
Pipeline Overview
Briefly, the rsvrecon pipeline performs the following major steps:
Merge Raw Reads (optional) Concatenate re-sequenced FastQ files (cat).
Quality Control of Raw Reads Evaluate sequencing quality (FastQC).
Adapter Trimming Remove adapters and low-quality bases (fastp).
RSV Database Mapping Map reads against RSV-specific databases (KMA).
Sequence Alignment
- Align reads to a reference (BWA(default) or STAR).
- Sort and index aligned sequences (SAMtools).
- Alignment quality assessment (SAMtools).
- Genome Assembly
- Calculate genome coverage (IGVtools).
- Perform reference-guided genome assembly (custom script).
Variant Identification Determine viral clade assignments, mutations, and sequence quality (NextClade).
Genotyping (Whole Genome & G-gene)
Pipeline Usage
[!NOTE] If youre new to Nextflow or nf-core, please refer to the nf-core installation guide. Run the workflow first with
-profile testto ensure proper functionality before applying it to your actual data.
Prepare your sample metadata in a CSV format (samplesheet.csv) with FASTQ files:
csv
sample,fastq_1,fastq_2
sample_1,sample_1_R1_001.fastq.gz,sample_1_R2_001.fastq.gz
sample_2,sample_2_R1_001.fastq.gz,sample_2_R2_001.fastq.gz
[!NOTE] If a sample has multiple sequencing lanes or replicates, list each replicate in a separate row with the same
sampleID. The pipeline will automatically merge these reads before analysis. Spaces in sample IDs will be automatically converted to underscores (_).
Run the workflow using the following command structure:
```bash
For St. Jude HPC users, specify the institutional profile, e.g., '-profile stjude'
nextflow run stjudecab/rsvrecon \
-profile
[!WARNING] Pipeline parameters should only be provided via CLI arguments or Nextflow's
-params-fileoption. Custom configuration files (-c) can specify system and pipeline configurations except for parameters. For detailed guidance, consult the configuration documentation.
For further information, please check the complete usage documentation and pipeline output descriptions.
Pipeline Output
Pipeline results are organized per sample ID within the specified output directory (<OUTDIR>), structured by analysis stages:
<OUTDIR>/<sample_id>/fastq: Contains trimmed FASTQ files ready for alignment.<OUTDIR>/<sample_id>/bam: Includes aligned BAM files and index files (BAI).<OUTDIR>/<sample_id>/qc: Quality control outputs for raw and aligned reads.<OUTDIR>/<sample_id>/reference: Reference genomes and database files used for the analysis.<OUTDIR>/<sample_id>/variant_calling: Variant calling and identification results.<OUTDIR>/<sample_id>/phylogeny_tree: Results related to phylogenetic analyses.<OUTDIR>/<sample_id>/assembly: Genome assemblies and coverage summaries.- (and more)
In addition to sample-level results, our pipeline delivers batch-level results as well, structured as follows:
<OUTDIR>/batch_qcs/: Contains MultiQC-based QC reports for both raw and filtered (trimmed) data.<OUTDIR>/batch_reports/: Include combined results across samples as clinical reports in bothPDFandHTMLformats.
Credits
stjudecab/rsvrecon was developed by Haidong Yi (@HaidYi) and Lei Li (@LeiLi-Uchicago) at the Center for Applied Bioinformatics (CAB), St. Jude Children's Research Hospital. The pipeline design incorporates community-driven best practices, especially inspired by nf-core. We also thank the wider CAB team for their valuable inputs and feedbacks.
![]()
Contributions and Support
We encourage community contributions. Please adhere to nf-core guidelines for maintaining consistency. Suggestions and improvements are welcome through pull requests or issues on our GitHub repository.
Disclaimer
The pipeline logo is initially generated through ChatGPT's
new 4o Image Generation function using the pipeline introduction as the prompt.
{kind=link}
Citations
bibtex
@article{Li2025.rsvrecon,
author = {Li, Lei and Yi, Haidong and Brazelton, Jessica N. and Webby, Richard and Hayden, Randall T. and Wu, Gang and Hijano, Diego R.},
title = {Bridging Genomics and Clinical Medicine: RSVrecon Enhances RSV Surveillance with Automated Genotyping and Clinically-important Mutation Reporting},
elocation-id = {2025.06.03.657184},
year = {2025},
doi = {10.1101/2025.06.03.657184},
publisher = {Cold Spring Harbor Laboratory},
URL = {https://www.biorxiv.org/content/early/2025/06/09/2025.06.03.657184},
journal = {bioRxiv}
}
Owner
- Name: St. Jude Center for Applied Bioinformatics (CAB)
- Login: stjudecab
- Kind: organization
- Location: Memphis, TN
- Website: https://www.stjude.org/research/shared-resources/center-for-applied-bioinformatics-cab.html
- Repositories: 1
- Profile: https://github.com/stjudecab
GitHub Events
Total
- Release event: 1
- Watch event: 1
- Public event: 1
- Push event: 3
- Pull request event: 1
Last Year
- Release event: 1
- Watch event: 1
- Public event: 1
- Push event: 3
- Pull request event: 1
Dependencies
- mshick/add-pr-comment b8f338c590a895d50bcbfa6c5859251edc8952fc composite
- actions/checkout 0ad4b8fadaa221de15dcec353f45205ec38ea70b composite
- conda-incubator/setup-miniconda a4260408e20b96e80095f42ff7f1a15b27dd94ca composite
- eWaterCycle/setup-apptainer main composite
- jlumbroso/free-disk-space 54081f138730dfa15788a46383842cd2f914a1be composite
- nf-core/setup-nextflow v2 composite
- actions/stale 28ca1036281a5e5922ead5184a1bbf96e5fc984e composite
- actions/setup-python 82c7e631bb3cdc910f68e0081d67478d79c6982d composite
- eWaterCycle/setup-apptainer 4bb22c52d4f63406c49e94c804632975787312b3 composite
- jlumbroso/free-disk-space 54081f138730dfa15788a46383842cd2f914a1be composite
- nf-core/setup-nextflow v2 composite
- actions/checkout 0ad4b8fadaa221de15dcec353f45205ec38ea70b composite
- actions/setup-python 82c7e631bb3cdc910f68e0081d67478d79c6982d composite
- peter-evans/create-or-update-comment 71345be0265236311c031f5c7866368bd1eff043 composite
- actions/checkout 0ad4b8fadaa221de15dcec353f45205ec38ea70b composite
- actions/setup-python 82c7e631bb3cdc910f68e0081d67478d79c6982d composite
- actions/upload-artifact 65462800fd760344b1a7b4382951275a0abb4808 composite
- nf-core/setup-nextflow v2 composite
- pietrobolcato/action-read-yaml 1.1.0 composite
- dawidd6/action-download-artifact bf251b5aa9c2f7eeb574a96ee720e24f801b7c11 composite
- marocchino/sticky-pull-request-comment 331f8f5b4215f0445d3c07b4967662a32a2d3e31 composite
- actions/checkout 0ad4b8fadaa221de15dcec353f45205ec38ea70b composite
- mshick/add-pr-comment b8f338c590a895d50bcbfa6c5859251edc8952fc composite
- nichmor/minimal-read-yaml v0.0.2 composite