lammpsstructures
Science Score: 62.0%
This score indicates how likely this project is to be science-related based on various indicators:
-
✓CITATION.cff file
Found CITATION.cff file -
✓codemeta.json file
Found codemeta.json file -
✓.zenodo.json file
Found .zenodo.json file -
○DOI references
-
✓Academic publication links
Links to: arxiv.org, rsc.org -
○Academic email domains
-
✓Institutional organization owner
Organization moss-bu has institutional domain (www.bu.edu) -
○JOSS paper metadata
-
○Scientific vocabulary similarity
Low similarity (16.5%) to scientific vocabulary
Repository
Basic Info
- Host: GitHub
- Owner: MoSS-BU
- Language: Python
- Default Branch: main
- Size: 76.4 MB
Statistics
- Stars: 0
- Watchers: 1
- Forks: 1
- Open Issues: 0
- Releases: 0
Metadata Files
README.md
LAMMPSStructures
Intro to the Extended Version
This package makes the setup of highly-customizable LAMMPS simulations of complex elastic structures simple. Its main purpose is to abstract away from the minutia of LAMMPS primitives to the FEA-inspired world of structures defined by nodes and connections between nodes. The source files are in the lammpspypack/lammpsWithPython directory: https://github.com/Keenan-Wood/LAMMPSStructures-1/tree/main/lammpspypack/lammpsWithPython.
This project was forked from the original LAMMPSStructures (https://github.com/adguerra/LAMMPSStructures), written by Arman Guerra. This extension provides a new, more general framework for defining elastic structures in LAMMPS, as well as direct integration with LAMMPS and the visualization software Ovito via python - amounting to a substantial overhaul. Changes made were designed with backwards compatibility in mind - scripts using the previous version should still run identically (albeit with deprecation warnings for certain replaced methods).
As a high-level overview, the lammpssimulation.py module contains a Simulation class processing lammps-specific data to create an input file (typically called in.mainfile) for lammps to run. The new runlammps() method can then be called to run the lammps simulation using that created file, and the output rendered programmatically with the lammpsrender.py module using ovito's python package. The new lammpsstructure.py module contains Structure, Node, Element, etc. classes to define large-scale frame-like elastic objects. Patterning methods and the ability to specify an element's shape using parametric equations make creating complicated geometry easy. Discretization methods then break the structure up into LAMMPS atoms connected by bonds, and the lammpssimulation.py methods can be applied directly to discretized structures.
The example and validation simulations mentioned in the original project's readme (below), the project file structure, and most of the lammpssimulation.py module are part of the original LAMMPSStructures project. The lammpsstructure.py, lammpsrender.py, lammpsbondstyles.py, and lammpsutils.py modules, as well as the addatoms(), applynodeconstraints(), addbondtypes(), addbonds(), and runlammps() methods in lammpssimulation.py are new additions, as well as the examples and validation simulations within the lammpsWithPython folder.
Getting Started & Reproducing Results
To get started, clone the repository, navigate to the env_setup folder, and in a terminal enter the commands:
conda env create -f environment.yaml conda activate lammps-env-1
This creates and activates a conda environment called 'lammps-env-1' - please edit the name of the environment in the environment.yaml file if there is already a different lammps-env-1.
To run the examples in LAMMPSStructures/examples, navigate to the directory and run the python script (ie. from examples/mushroom, enter the command 'python mushroom.py mushroomsim1'). Some examples, like this mushroom script, require an argument for the simulation name - a folder with this name will be created to hold simulation files.
To run the examples using the new lammpsstructure.py module, navigate to the lammpspypack/lammpsWithPython/examples directory (https://github.com/Keenan-Wood/LAMMPSStructures-1/tree/main/lammpspypack/lammpsWithPython/examples/) and simply run the python script corresponding to the chosen example (ie. run the command 'python helixarray.py').
Example code
The following examples showcasing the new lammpsstructure.py functionality are located in lammpspypack/lammpsWithPython/examples (https://github.com/Keenan-Wood/LAMMPSStructures-1/tree/main/lammps_pypack/lammpsWithPython/examples/).
Simple Column Buckling
testsimplebeam.py and the testsimplebeam folder
This simple tutorial shows how to setup a simple simulation of a long slender column under compression. The testsimplebeam folder with all of the simulation files is generated by the lammps_simulation.py module.

Cantilevered Truss
truss.py and the truss folder
This example showcases the power of the linear patterning and transformation features. First, a unit-cell is created, in this case a square frame with a cross member:

Next, the unit cell is translated so that the final truss will be centered on the origin:
new_structure.translate([-d_between_beams*n_beams/2, 0, -beam_length/2], copy=False)

The unit cell is patterned along the x-axis once, to allow for the application of bonds between elements through nodes along the length (which will be applied to connect future copies as well):
new_structure = new_structure.pattern_linear(np.array([1,0,0]), 1, offset = d_between_beams)

Finally the dual-cell is patterned along the x-axis the number of times specified:
new_structure = new_structure.pattern_linear(np.array([1,0,0]), n_beams - 3, offset = d_between_beams)

All of this works by defining affine transformations on a structure's coordinates, as well as by defining addition for two structures to join them logically.
Helix Array
helixarray.py and the helixarray folder
This examples shows how straightforward it is to define curved elements between nodes by parametric equations. Here is an excerpt, showing the parametric equation definition with lambda functions. The parameter t is taken to be from 0 at the element's nodea to 1 at the element's nodeb, and the value of the parametric equations should be 0 at nodea, and nodeb.coords - nodea.coords at nodeb.
``` nodediameter = beamthickness nodes = [ ([0, 0, 0], nodediameter), (None, nodediameter), ([dbetweenbeams, 0, 0], nodediameter), (None, nodediameter) ] constraints = [[1, 1,1,1], [3, 1,1,1]]
(E, rho) = (Ebeams, density) materials = [['testmaterial0', E, rho]] xsecs = [[0, beamthickness]]
rhelix = 0.004 Nturns = 15 parameqs = [lambda t: rhelix(np.cos(N_turns2np.pit) - 1), lambda t: rhelix*np.sin(Nturns2np.pit), lambda t: -beam_lengtht] elements = [(0, 1, parameqs), (2, 3, parameqs), (0,2)]
newstructure = lstruct.Structure( nodelist = nodes, materiallist = materials, xsectionlist = xsecs, elementlist = elements, constraintlist = constraints) ```


Validation
The proper choice of additional bond parameters ought to be justified on a case-by-case basis. Here basic 2-atom bonds and 4-atom dihedral potentials are applied by default - agreement with axial stretching and classic beam-bending formula validate their use. To hold the structure together, default bonds are applied based on the specified material and xsection properties:
A harmonic two-atom bond potential is used for axial stiffness:

https://docs.lammps.org/bond_harmonic.html
The default stiffness applied is K = Young's Modulus * atomdiameter*2 / (2restlength), as validated in the original LAMMPSStructures. This bond is applied to adjacent atoms in every element, as well as to end-element atoms and the atom of a neighboring node.
An 'angle cosine delta' three-atom bond is defined for adding bending stiffness between element pairs at nodes:

https://docs.lammps.org/anglecosinedelta.html
By default this is not applied internally - the bending rigidity is included in the dihedral spherical potential for numeric stability. The default angular stiffness, when applied between elements at nodes, is K = Young's Modulus * atomdiameter*4 / (12restlength), as validated in the original LAMMPSStructures.
A 'dihedral spherical' four-atom bond adds bending rigidity and out-of-plane bending stiffness for parametricly-defined elements:

https://docs.lammps.org/dihedral_spherical.html
The default potential applied to enforce bending rigidity follows the form of the second example in the lammps documentation:


The rest planar angles and rest dihedral angle are calculated from the atom coordinates, and the same angular stiffness as for the angle cosine delta potential is used for the planar angle stiffness. As a basic default, the dihedral stiffness is set to 0.2 times this angular stiffness for curved elements, and 0 for straight elements. Since this increases the energy in the system, greater viscosity is needed in some cases for convergence.
Current work lies in not only validating the correct creation of the lammps input file, but in also fine-tuning the default dihedral stiffness parameter to more closely model the behavior of curved beams.
ME 700 - Applied Skills
● Github project structuring and management
● Coding environment setup and automation (bash script)
● Modular, object-oriented programming in python
● Thorough exception handling - Writing robust code
● Useful and concise documentation with standard formatting
● Analyzing and expanding upon code written by others
● Interfacing with large open-source simulation software
● Simulation validation using problems with analytical solutions
● Debugging strategies (particularly using VS Code)
● Standard FEA geometry setup - ie. definition with nodes, elements, materials, etc.
End Extended Version readme
The following sections of the readme is left unchanged from the original LAMMPSStructures, for easy reference. Future consolidation will see these sections merged. Current work includes implementing module testing with pytest, fixing dihedral potential numerical stabillity issues, and expanding on tutorials (and converting to jupyter notebooks).
Intro
The goal of this repository is to document and provide some examples for a python package which facilitates the use of LAMMPS as a simulation tool for elastic materials. This python package does not simulate things on its own, rather, it contains a class and functions that allow you to write a set of files which can then be run with LAMMPS.
Elastic structures are traditionally simulated using a finite element software, which has the benefit that it can model complex geometries. However, often these softwares have a very hard time modeling contact and other complex boundary conditions. For some examples, when roots dig into soil, when cells jam on the surface of the Extra-Cellular Matrix, or when birds build a nest, there is a coupling either between many elastic bodies in contact, or between granular and elastic materials. These would all be very hard to simulate using a finite element software.
On the other hand, LAMMPS, a Discrete Element simulation software, is excellent at handling contact at the expense of geometrical complexity -- almost everything is treated as a sphere. In a couple of recent papers [1,2,3,4,5], I and others have shown that it is possible to get the best of both worlds. By "gluing" spheres together to form some simple geometries that can be endowed with elastic properties, we can have contact and elasticity in the same simulation. This repository, I hope, should contain everything you need to start doing this yourself.
Brief Tour
We have three main directories in this repo:
lammps_pypack: This contains the python package that we will use to make the files that will allow us to simulate elastic and granular materialsexamples: These are some examples of this package being used in action. Some of these examples are systems that we have studied in specific publications, but others are just examples that we thought would be intersting for future study, or which display the capacity of this tool.validation: Here there are some simulations which we validate either theoretically or against other simulations in COMSOL. This directory is comparatively scant, just because we have also performed validation as part of some of the studies cited earlier which use this tool 1,2,3,4,5
There are also some files which you might find useful:
Documentation.md: A brief description of all of the functions available in thelammpsWithPython.lammps_objecttool. More thorough descriptions are given in the actual function definitionsCITATION.cff: You can cite this if you use this tool in an academic setting. If you use simulations that are similar to any of the simulations in these publications 1,2,3,4,5, then cite the publication as well.
## How to use this tool
To begin to use this tool, clone this repository and pip install lammpsWithPython from within the lammps_pypack folder. You can then run an example file, or make a new file which sets up some simulation that you make.
Any time you use this tool, it will create a new folder with a name that you specify, and within that folder, it will write some files that LAMMPS will need to run a simulation. It will always write a file called in.main_file, which is the main input LAMMPS file that you will eventually run. This sets up the simulation, determines the dimensionality and size of your simulation box, etc. The tool may also create some secondary files which will be accessed by in.main_file. For example, if you want to insert a grain (a sphere) in your simulation box, you will call the add_grains command, and input to that command, among other things, the coordinates of those grains. When you do that, a new file will be written, called grains_*.txt where the asterisk is some integer (namely, 1 if this is the first time you have inserted grains, 2 if it is the second, etc.). Calling the add_grains command also writes a line to in.main_file (include grains_*.txt) which makes LAMMPS look in the grains_*.txt file for the locations of the grains. You therefore have to keep all of these files together, which is why they are all written together in a new folder every time you use the tool.
This multiple files business may seem complicated at first, but I hope that after you use this tool a couple of times, you'll appreciate that it creates a cleaner version of in.main_file, since long lists of particles and bonds are stored in other files.
To run the in.main_file of any of these simulations, you will first need to download and install LAMMPS. Once you have LAMMPS installed, you can go into the folder which contains in.main_file and input it to LAMMPS (something like lmp_serial -i in.main_file, this will depend on how lammps is built on your machine). This tool is currently set to run with the LAMMPS version which was released on June 23, 2022, and so if something doesn't work, let me know and I can try to adjust it! Or adjust it yourself (thats what Git is for right??).
Some Other Examples
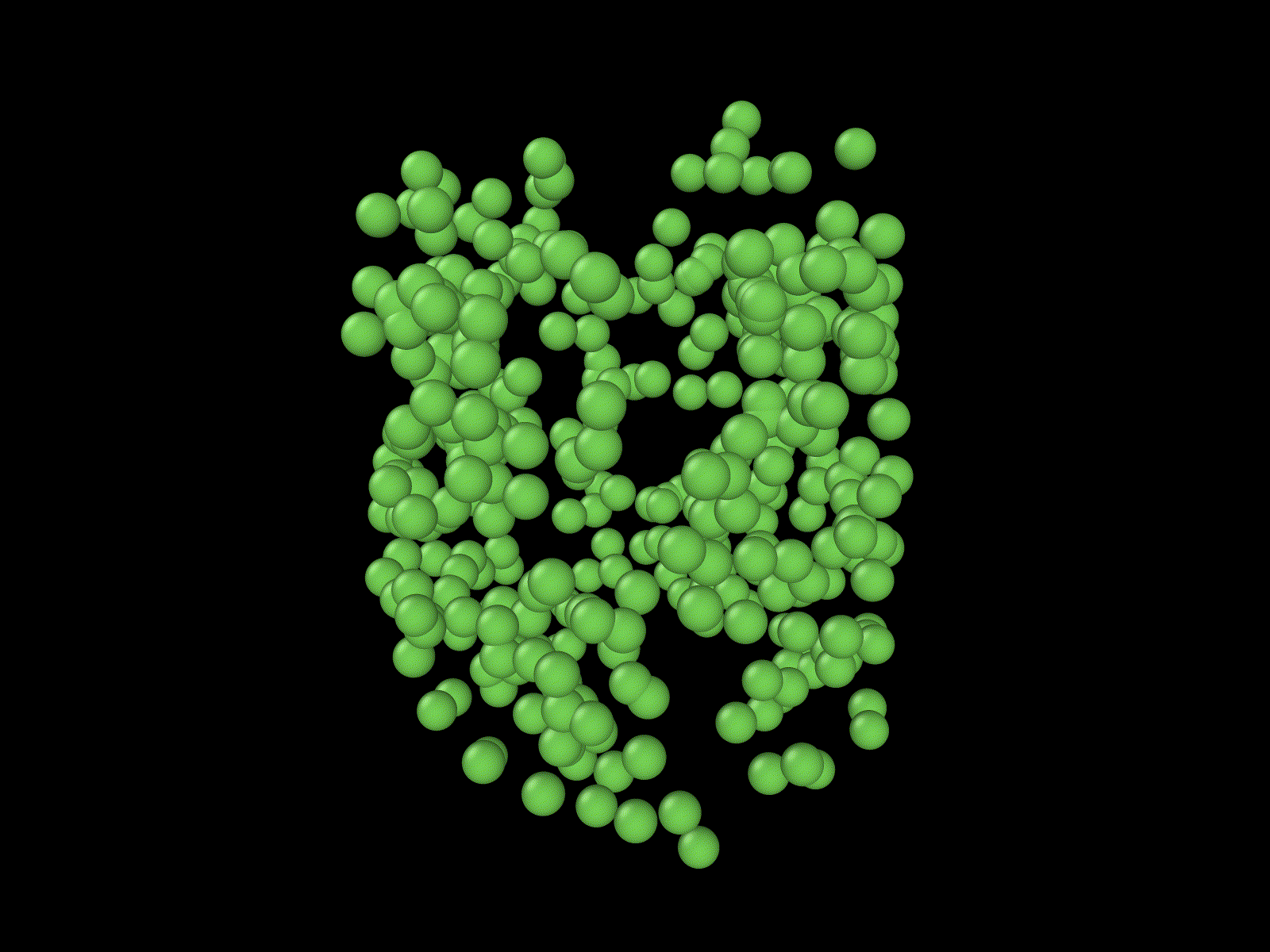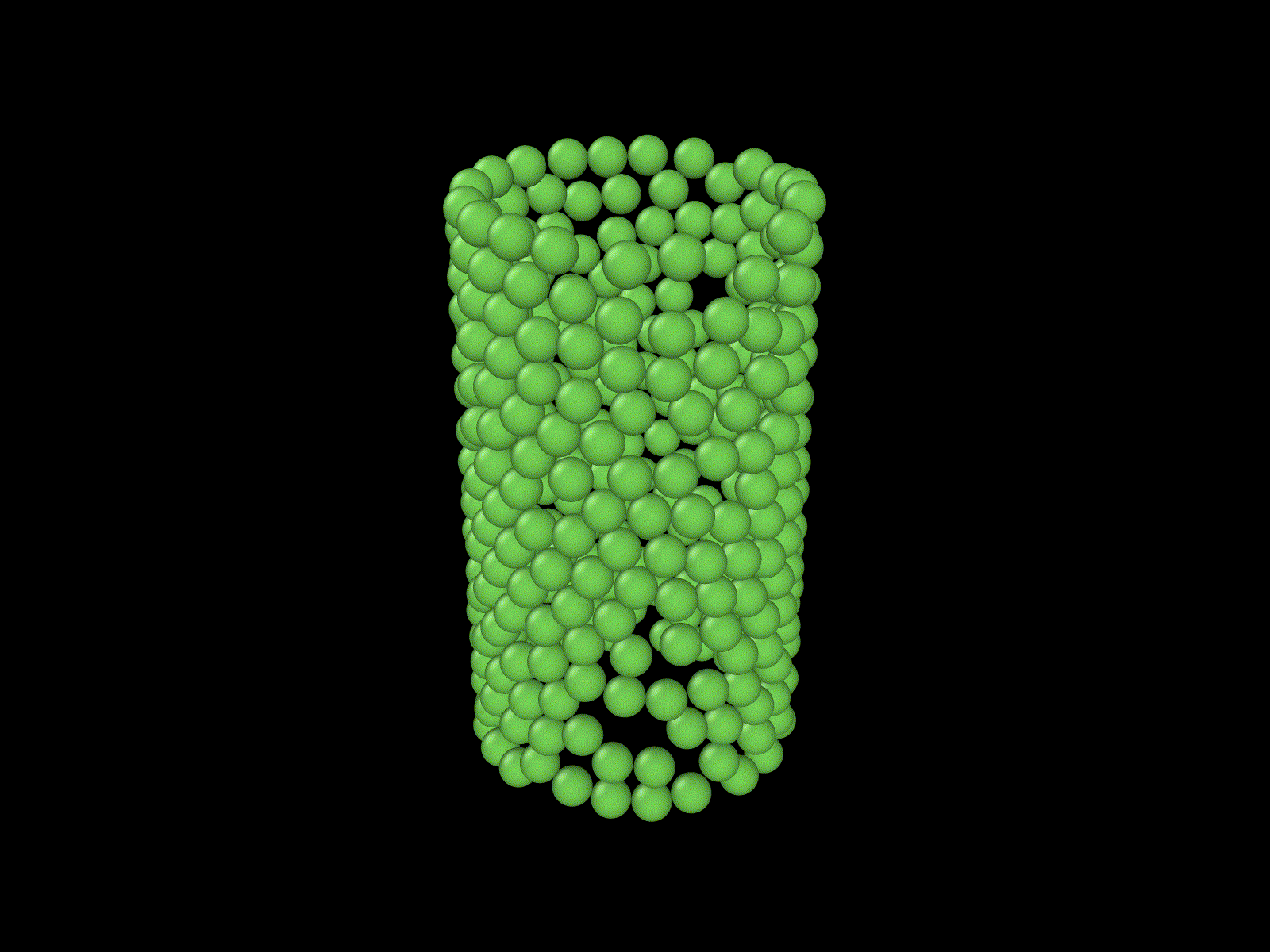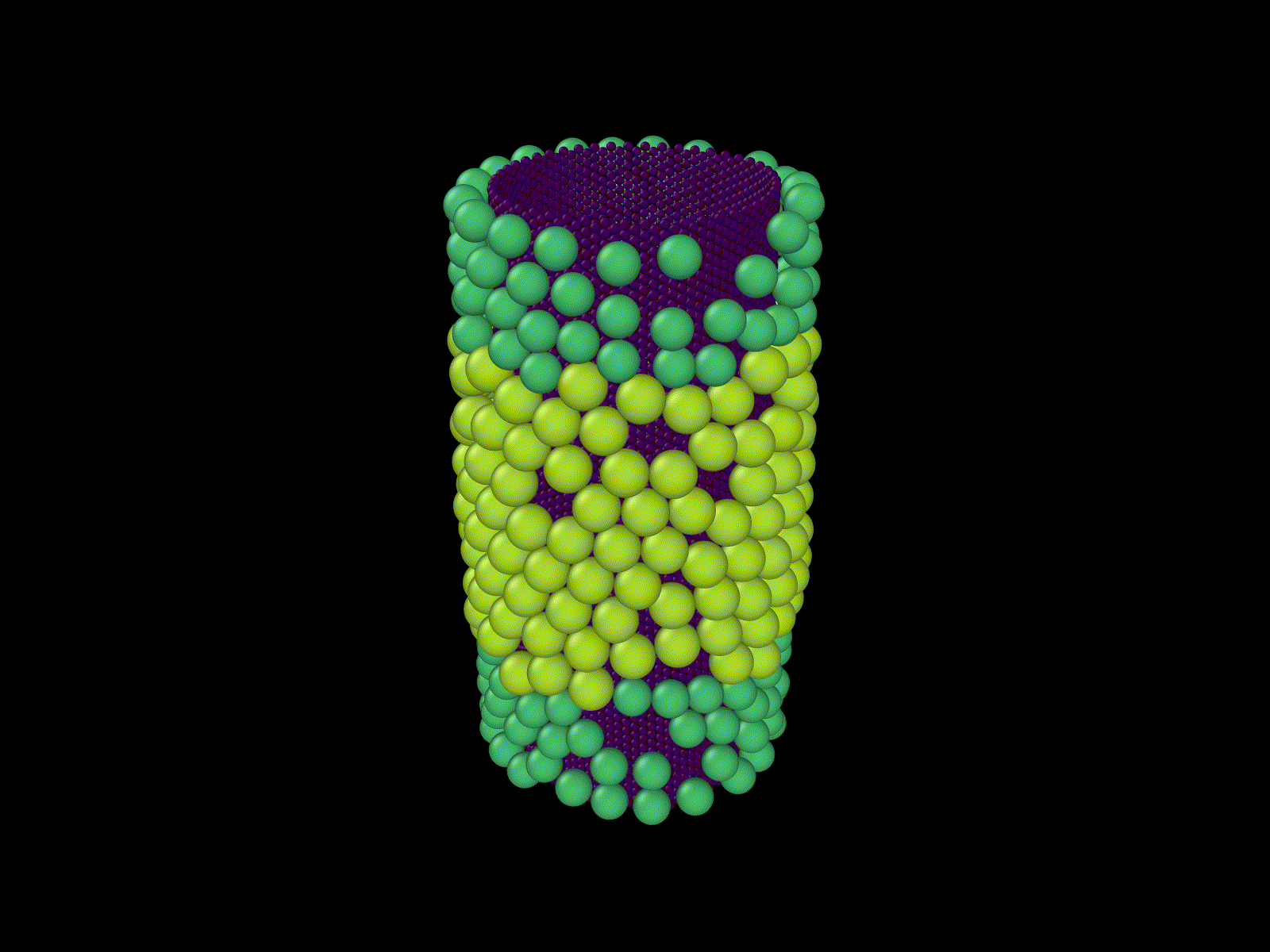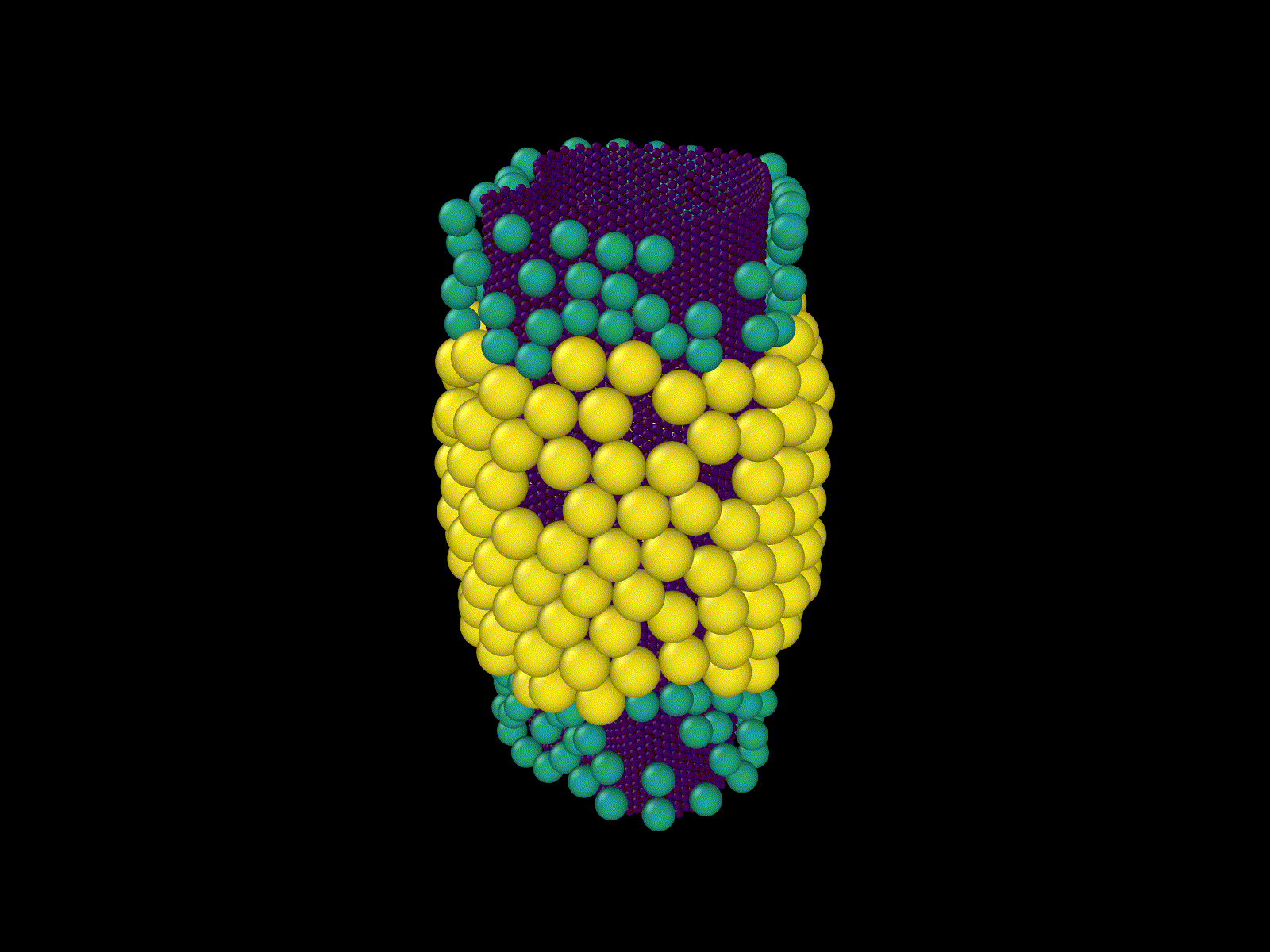
These examples show some simulations that I do not have examples of here, but that I thought were cool.
"Cells" growing on a manifold
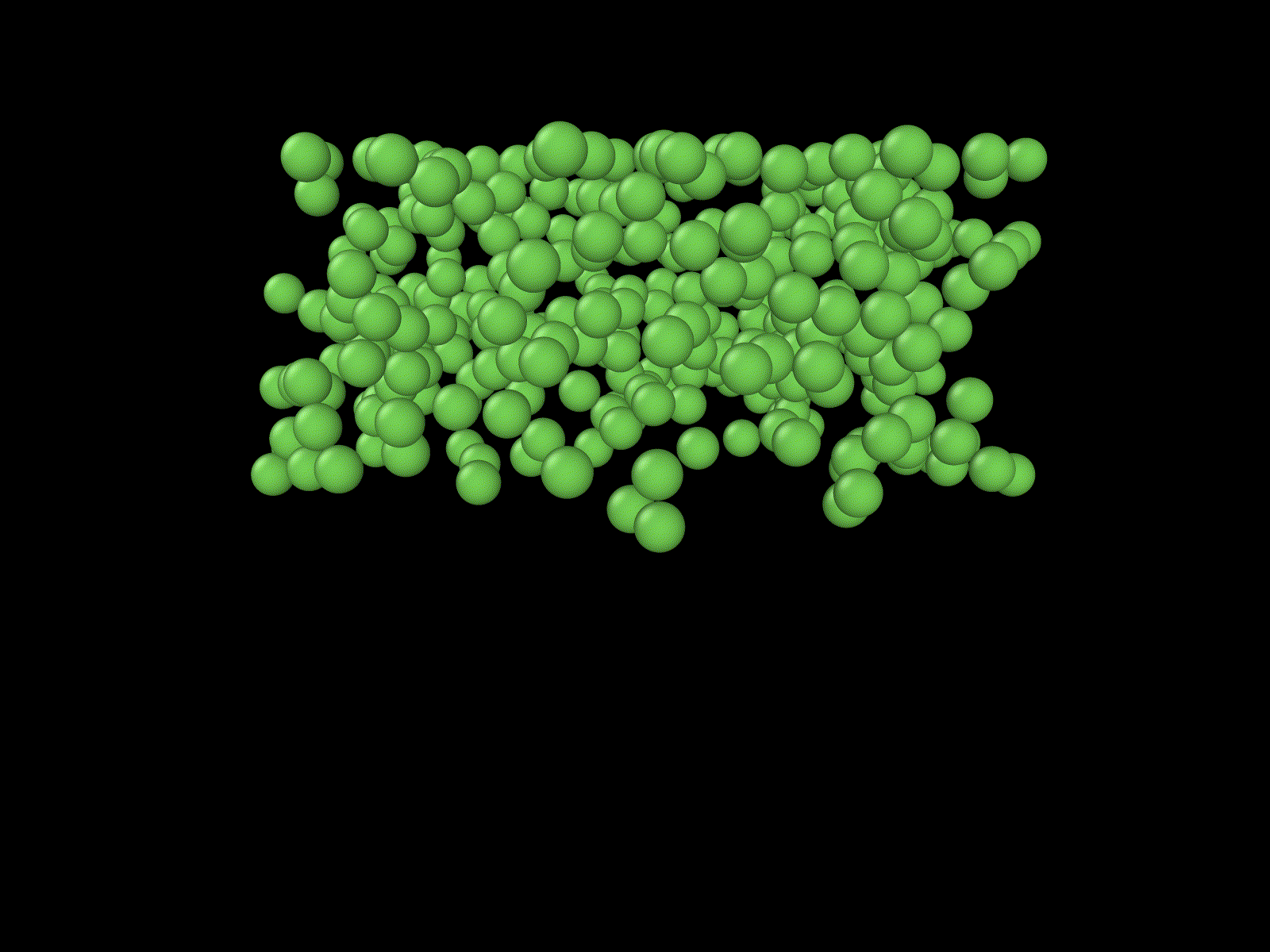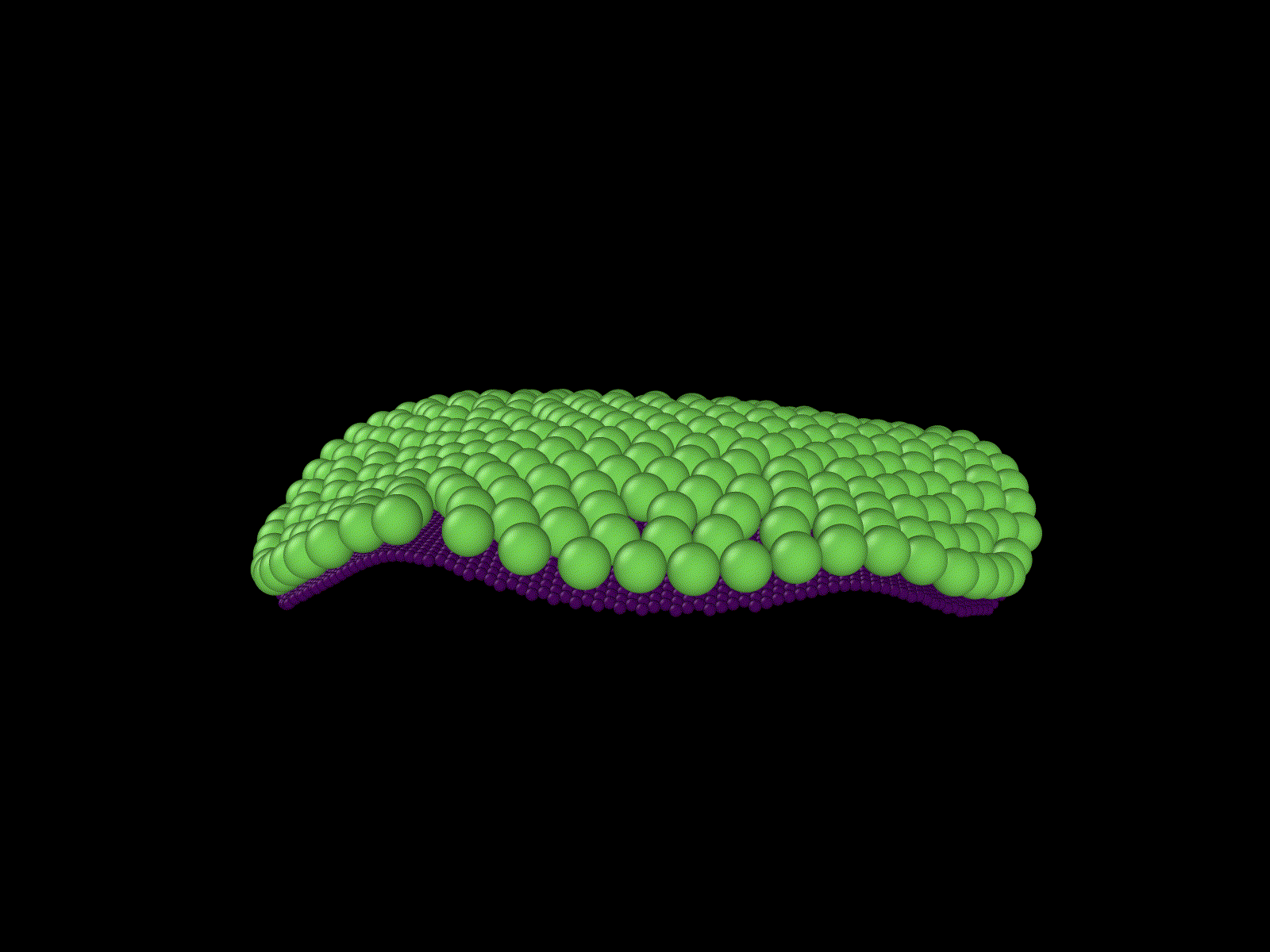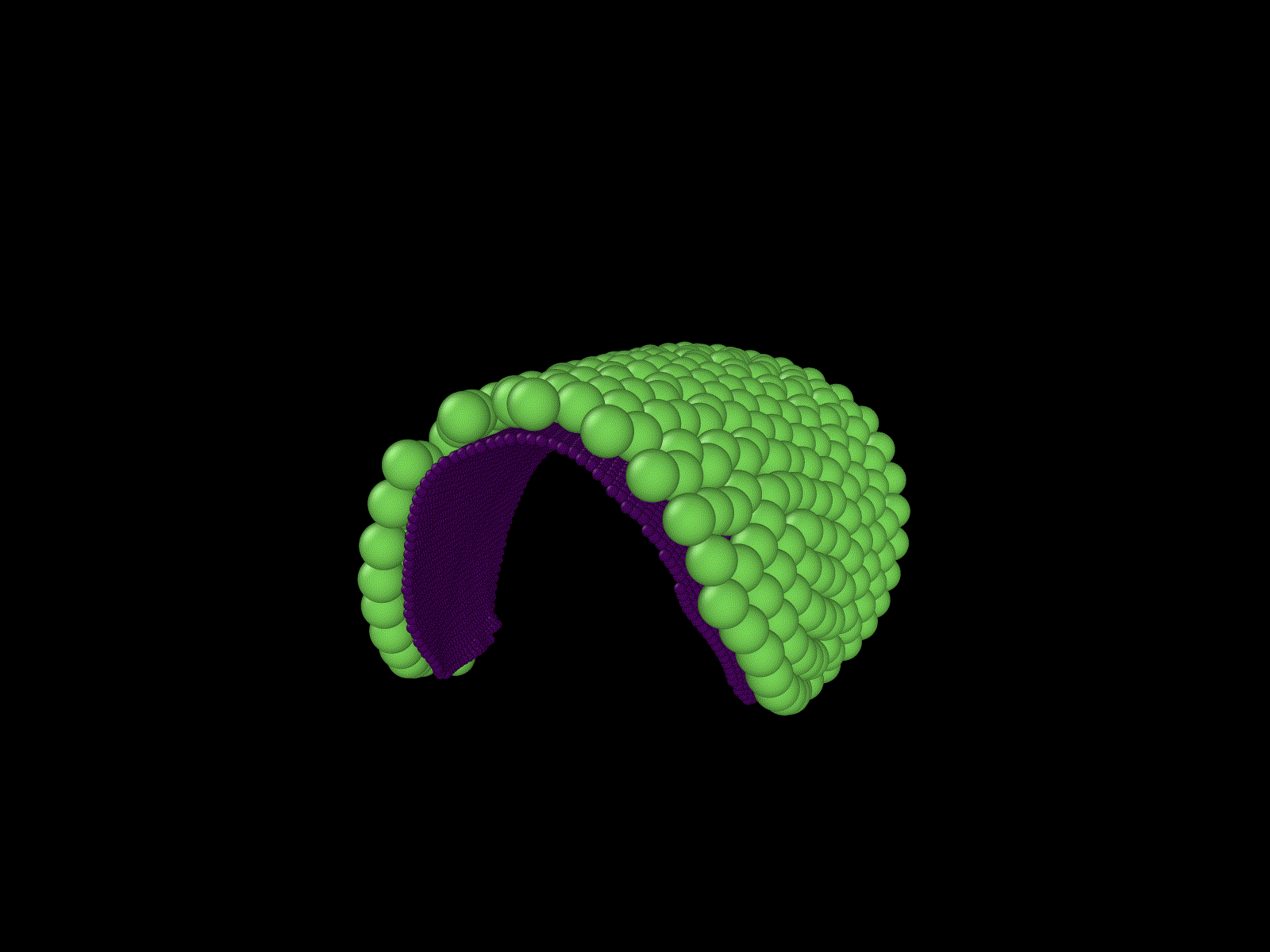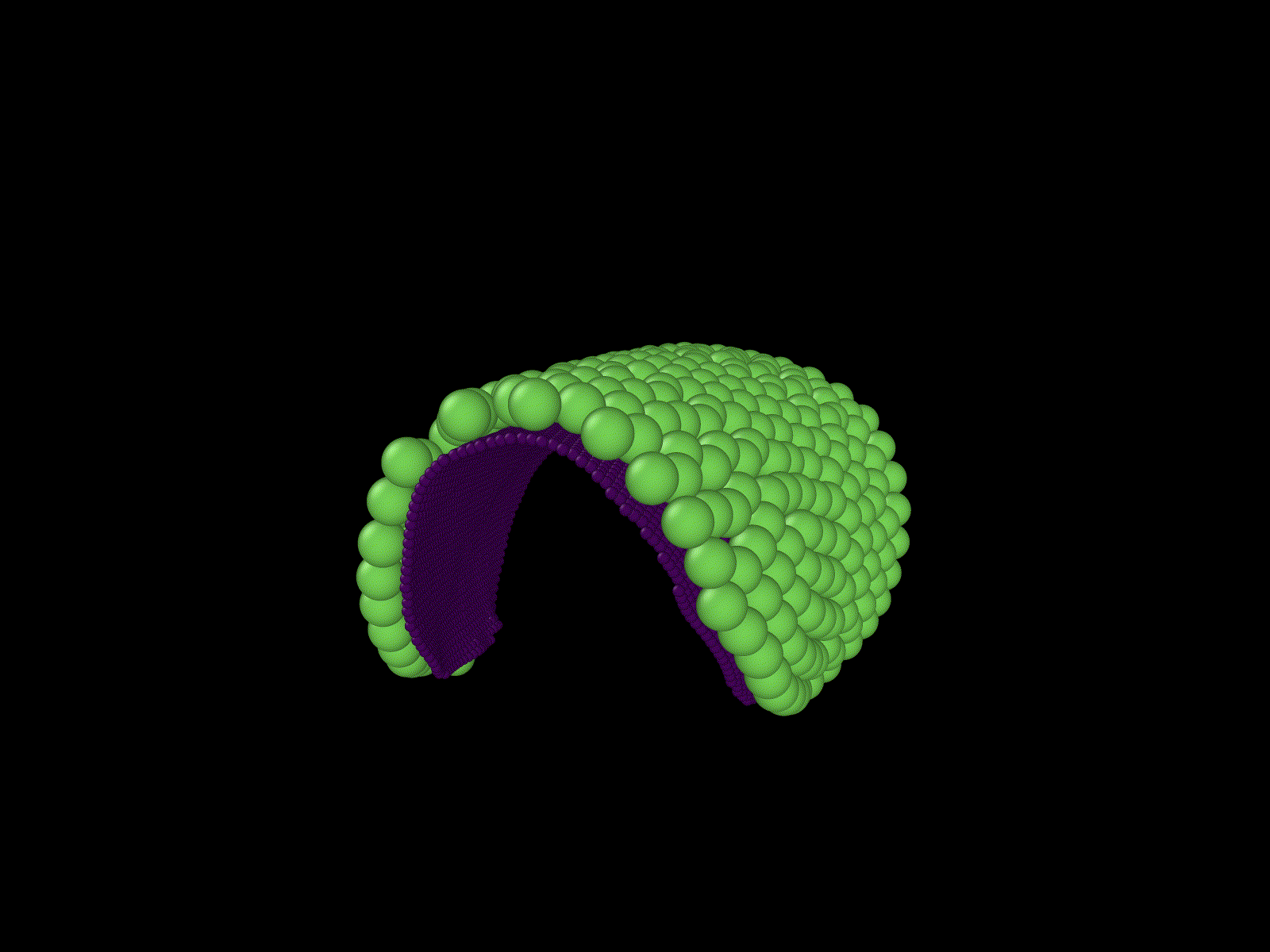
Simulation of a knot

Frustrated growth on a cylinder
Owner
- Name: Mechanics of Slender Structures
- Login: MoSS-BU
- Kind: organization
- Email: dpholmes@bu.edu
- Website: http://www.bu.edu/moss/
- Repositories: 1
- Profile: https://github.com/MoSS-BU
The MoSS Lab at Boston University studies how objects change shape.
Citation (CITATION.cff)
# This CITATION.cff file was generated with cffinit.
# Visit https://bit.ly/cffinit to generate yours today!
cff-version: 1.2.0
title: LAMMPSStructures
message: >-
If you use this software, please cite it using the
metadata from this file.
type: software
authors:
- given-names: Arman
family-names: Guerra
email: adguerra@bu.edu
affiliation: Boston University
orcid: 'https://orcid.org/0000-0001-7813-4955'
date-released: '2022-04-22'
GitHub Events
Total
- Push event: 28
- Pull request event: 1
- Create event: 2
Last Year
- Push event: 28
- Pull request event: 1
- Create event: 2