


A reproducible Snakemake pipeline to analyse Illumina paired-end data from ChiP-Seq experiments
A reproducible Snakemake pipeline to analyse Illumina paired-end data from ChiP-Seq experiments - Published in JOSS (2019)

catd_snakemake
Snakemake pipeline for benchmarking cell-type deconvolution methods and deconvolving real bulk RNA-seq data with the use of scRNA-seq datasets


snakemake-workflow-fermi-lat
A Snakemake workflow for Fermi-LAT data reduction

metagem
:gem: An easy-to-use workflow for generating context specific genome-scale metabolic models and predicting metabolic interactions within microbial communities directly from metagenomic data

dea_limma
A Snakemake workflow and MrBiomics module for performing and visualizing differential (expression) analyses (DEA) on NGS data powered by the R package limma.
spilterlize_integrate
A Snakemake workflow and MrBiomics module to split, filter, normalize, integrate and select highly variable features of count matrices resulting from next-generation sequencing (NGS) experiments (e.g., RNA-seq, ATAC-seq, ChIP-seq, Methyl-seq, miRNA-seq,...) including confounding factor analysis and diagnostic visualizations.
mixscape_seurat
A Snakemake workflow and MrBiomics module for performing perturbation analyses of pooled (multimodal) CRISPR screens with sc/snRNA-seq read-out (scCRISPR-seq) powered by the R package Seurat's method Mixscape.
dea_seurat
A Snakemake workflow and MrBiomics module for performing differential expression analyses (DEA) on (multimodal) sc/snRNA-seq data powered by the R package Seurat.

euryale
A pipeline for taxonomic classification and functional annotation of metagenomic reads. Based on MEDUSA


snhic
Snakemake pipeline for analysis and normalization of Hi-C data starting from fastq.gz files. It includes the possibility to perform grouped analyses, TAD, loops and stripes detections, as well as differential compartment and chromatin interaction analyses.
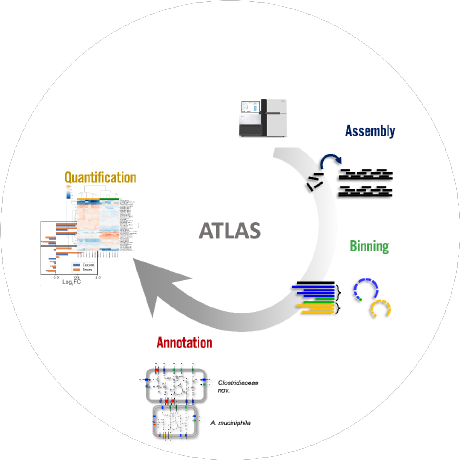
metagenome-atlas
ATLAS - Three commands to start analyzing your metagenome data

vipera
A Snakemake workflow for SARS-CoV-2 Viral Intra-Patient Evolution Reporting and Analysis

transcluster
A Snakemake workflow to find SARS-CoV-2 clusters in a reference phylogeny and estimate their normalized clustering index

scanneo2
Snakemake-based computational workflow for neoantigen prediction from diverse sources
unsupervised_analysis
A general purpose Snakemake workflow and MrBiomics module to perform unsupervised analyses (dimensionality reduction & cluster analysis) and visualizations of high-dimensional data.
https://github.com/adonath/snakemake-workflow-chandra
A Snakemake workflow for Chandra data reduction


solar-and-wind-potentials
Estimation of solar and wind power generation potentials in Europe.

https://github.com/cbg-ethz/v-pipe
V-pipe is a pipeline designed for analysing NGS data of short viral genomes
https://github.com/cbg-ethz/pybda
:computer::computer::computer: A commandline tool for analysis of big biological data sets for distributed HPC clusters.

snakemake
This is the development home of the workflow management system Snakemake. For general information, see

https://github.com/biocore/qadabra
Snakemake workflow for comparison of differential abundance ranks

https://github.com/cbg-ethz/sars-cov-2_analysis
A Snakemake workflow for large-scale SARS-CoV-2 analyses.

https://github.com/charlesfoster/mitowrap
A snakemake pipeline wrapping MitoZ and getOrganelle for de novo mitogenome assembly using short reads and subsequent QC.


https://github.com/clavellab/genome-assembly
A Snakemake workflow assembling bacterial genomes according to the standard operating procedure in the Clavel Lab

sv-gen
Snakemake-based workflow for generating artificial genomes with structural variants

fair_bowtie2_mapping
Align reads over a reference genome, filter aligned-reads, and mark duplicates

snakemake_rnaseq
A Snakemake pipeline to go from fastq mRNA sequencing files to raw and normalised counts (usable for downstream EDA and differential analysis)
fair_gatk_mutect2
Snakemake workflow used to call germline and/or somatic variants with GATK Mutect2

viroconstrictor
ViroConstrictor is a pipeline designed to process raw FastQ data from viral amplicon-based sequencing and generate biologically correct consensus sequences of the given viral genome


snakemake-novice-lattice
Introduction to Snakemake for Lattice Quantum Field Theory
genome_tracks
A Snakemake workflow and MrBiomics module for easy visualization of genome browser tracks of aligned BAM files (e.g., RNA-seq, ATAC-seq, scRNA-seq, ...) powered by the wrapper gtracks for the package pyGenomeTracks, and IGV-reports.

snakemake-publishing
Lessons on taking a basic Snakemake workflow and sharing it with the community

cnakepit
A Snakemake pipeline for copy number variant calling without normal tissue samples

megaisurv-namaste
Nanopore Metagenomic Antibiotic Resistance and Taxonomy Screening

https://github.com/angrymaciek/mapp
🗺️ MAPP is a computational method which enables identification of binding motifs for RNA-binding proteins that shape pre-mRNA processing under specific conditions.
mpox-seek
A rapid pipeline for targeted and whole-genome ONT monkeypox sequencing
pypsa-eur
PyPSA-Eur: A Sector-Coupled Open Optimisation Model of the European Energy System
scrnaseq_processing_seurat
A Snakemake workflow and MrBiomics module for processing and visualizing (multimodal) sc/snRNA-seq data generated with 10X Genomics Kits or in the MTX matrix file format powered by the R package Seurat.

grenepipe
A flexible, scalable, and reproducible pipeline to automate variant calling from raw sequence reads, with lots of bells and whistles - for sampled individuals, and for pool sequencing.

spikeflow
Pipeline to analyse ChIP-Rx data, i.e ChIP-Seq with reference exogenous genome spike-in normalization
zarp-cli
A user-friendly command-line interface for the ZARP RNA-seq analysis workflow
ena-spike-ntd-repdel-analysis
A Snakemake workflow with associated scripts used for detecting spike NTD repaired deletions in SARS-CoV-2 Omicron BA.1 lineage reads. Manuscript under review.
sv-callers
Snakemake-based workflow for detecting structural variants in genomic data
https://github.com/angrymaciek/warlock
Warlock is a snakemake workflow to spawn multiple demons (deme-based oncology models) as jobs running around on a cluster environment 😈😈


popglen
Bioinformatics pipeline to process whole genome resequencing data and perform genotype likelihood based population genomic analyses using ANGSD and related softwares. Flexible to datasets that combine high/low coverage and historical/fresh samples.
fair_genome_indexer
Download and index Ensembl sequences and annotations, remove non-canonical chromosimes, remove low TSL, index with multiple tools

tucca-rna-seq
Tufts University Center for Cellular Agriculture's RNA-Seq Workflow for Cellular Agriculture Projects
enrichment_analysis
A Snakemake workflow and MrBiomics module for performing genomic region set and gene set enrichment analyses using LOLA, GREAT, GSEApy, pycisTarget and RcisTarget.


population-structural-var-calling-smoove
population structural variant calling with smoove
rnaseq_pipeline
RNA-seq Data Processing, Quantification and Annotation Snakemake Workflow and MrBiomics Module.

rna-seq-salmon-deseq2
Snakemake workflow for RNA-Seq differential transcript analysis using Salmon/Deseq2
atacseq_pipeline
Ultimate ATAC-seq Data Processing, Quantification and Annotation Snakemake Workflow and MrBiomics Module.
https://github.com/cbg-ethz/scdna-pipe
Python data analysis pipeline for single cell copy number event history reconstruction

rdscan
A snakemake workflow for regions of difference discovery in Mycobacterium tuberculosis complex (MTBC) samples


https://github.com/ccbr/ccbr_snakemaketemplate
Barebones framework for creating new snakemake workflows.